细胞内mRNA翻译影响因素及翻译组学的研究进展
2023-01-05马荣尚方正潘剑锋戎友俊王敏李金泉张燕军
马荣 尚方正 潘剑锋 戎友俊 王敏 李金泉 张燕军
(1.内蒙古农业大学动物科学学院,呼和浩特 010018;2.农业农村部肉羊遗传育种重点实验室,呼和浩特 010018;3.内蒙古自治区动物遗传育种与繁殖重点实验室,呼和浩特 010018;4.内蒙古自治区山羊遗传育种工程技术研究中心,呼和浩特 010018)
蛋白质是基因的功能产物,是细胞功能的主要执行者,在发育、神经发生、记忆形成和衰老等多种生理过程中起着关键作用[1-2]。mRNA 是蛋白质合成的信息蓝图,基于高通量测序的转录组测序可以反映细胞中mRNA 的种类和数量,且可以检测mRNA 的序列突变和可变剪切[3-4]。Maier 等[5]对多个物种的转录组和蛋白组进行比较后发现,mRNA 表达水平与其编码蛋白质的表达量的相关系数在0.01-0.5 之间,表明mRNA 的表达水平并不能较好地作为判断蛋白质水平的依据。因此,作为转录组和蛋白组之间的“桥梁”——翻译调控被认为是调节基因表达的主要机制。翻译调控是一种快速激活或抑制mRNA 翻译以响应内源、外源信号的机制,可快速灵活的调节细胞内蛋白质浓度,实现生理变化、维持体内环境稳态。翻译调控紊乱会导致细胞无法维持正常的细胞功能或无法适应不断变化的环境条件,从而导致多种疾病[6-7]。翻译可分为3个阶段:起始、延伸、终止。其中,翻译起始长久以来被认为是翻译速率的主要限速步骤[8],是决定蛋白质表达量的关键,影响着蛋白质的结构和功能,在细胞增殖、凋亡等方面扮演重要角色[9]。
细胞的生长速率、蛋白质合成速率都与核糖体数目密切相关。生物在不同生长阶段、不同生长环境以及应激条件下都是通过对翻译速率的调控实现对外界刺激迅速产生反应。翻译速率的探究有助于科研人员更清晰地了解细胞生命活动的快速变化。因此,本文在概述真核、原核细胞内翻译机制的基础上,从影响翻译速率的主要因素和探究翻译、翻译速率的主要技术手段两个方面综述了mRNA 翻译的研究现状,旨在为更多学者进一步了解生物体内翻译过程提供参考依据。
1 mRNA 翻译机制
核糖体是mRNA 进行翻译的主要场所,主要由核糖体RNA(ribosomal RNA,rRNA)和多种核糖体蛋白构成,是一类高度复杂的细胞器。核糖体内部有3 个转运RNA(transfer RNA,tRNA)结合位点,分别为E、P、A 位点,A 是氨酰-tRNA 结合的位点,P 是肽酰-tRNA 结合的位点,E 是核糖体内空载tRNA 释放位点。携有氨基酸的tRNA(氨酰-tRNA)会依次经过A、P 位点,随后将氨基酸结合于多肽链末端,最后以空载tRNA 的形式从E 位点移出[10]。当核糖体遇到终止密码子时,mRNA 翻译结束,多肽形成[10]。以上是细胞内mRNA 的翻译过程,分为3 个阶段:起始、延伸和终止。生物体内细胞的翻译过程是高度保守的[11],机制也十分相似。然而,由于真核、原核细胞内mRNA 结构的不同,因此两类细胞的翻译起始方式有明显差异,但两类细胞内的翻译延伸、终止是相似的[12]。因此本文将分别介绍真核细胞和原核细胞内的翻译机制。
1.1 真核生物mRNA翻译机制
真核细胞内存在两类核糖体识别机制,一类为5′端帽子识别机制,另一类为识别核糖体进入位点(internal ribosome entry site,IRES)机制。有研究表明,细胞内存在部分RNA(大多为非编码RNA,如lncRNA、circRNA 等)是通过对IRES 的识别进行翻译,但真核细胞内大多数mRNA 都是通过对5′端帽子结构的识别进行翻译。因此,本文以5′端帽子识别机制为例进行简要介绍。
首先,由支架蛋白eIF4G 和结合蛋白eIF4E 共同识别mRNA 的5′帽子结构。随后在eIF4G 因子的招募下,eIF4A 与mRNA 结合,eIF4A、eIF4E 和eIF4G 共同构成eIF4F,并在eIF4B 的共同作用下破坏mRNA 5′端的二级结构(如发夹结构等),这为接下43S 前起始复合物的结合提供条件[13]。与此同时,核糖体40S 小亚基也为mRNA 的结合做准备。为使与eIF2-GTP 结合的甲硫氨酸起始子tRNA(MettRNAi)顺利的结合在小亚基的P 位点,翻译起始因子eIF1 首先结合于小亚基E 点,随后eIF3、eIF5 依次与E 位点的eIF1 结合,共同占据E 位点;而小亚基A 位点则由翻译起始因子eIF1A 占据。翻译起始因子eIF5、eIF3、eIF1 和eIF1A 不仅可以确保MettRNAi 准确结合于核糖体P 位点,而且可以阻止核糖体大、小亚基的结合,进而确保翻译的顺利进行。eIF2 以GTP 的形式(eIF2-GTP) 与Met-tRNAi 结合,并在eIF5 的作用下将Met-tRNAi 迁至小亚基P点。此时,由核糖体40S 小亚基、eIF3、eIF5、eIF1和eIF1A 以及GTP、eIF2 和Met-tRNAi 构成的复合物为43S 前起始复合物。eIF3(位于43S 前起始复合物内)被结合于mRNA 5′端的eIF4G 招募,从而形成48S 前起始前复合物[13-14];48S 前起始前复合物从5′→ 3′方向扫描,直至找到具有Kozak 序列特征的起始密码子[15-16]。正确的碱基互补配对改变了48S 前起始复合物的构象,促使翻译起始因子从复合体中释放,核糖体大亚基与小亚基结合。此时,mRNA 翻译起始结束,翻译延伸开始。
在翻译延伸阶段,起始tRNA(Met-tRNAi)停留在具有延伸能力的核糖体的肽基(P)部位,而氨酰-tRNA 在延伸因子的“护送”下移至核糖体A 位点,A 位点氨酰-tRNA 和P 位点的氨基酸相遇并形成肽键。肽键形成后,核糖体移至下一密码子,P 位点的tRNA 从E 位点排出,A 位点的tRNA 带着合成的肽链移至P 位点,而A 位点空出,等待下一个氨基酸。翻译延伸的过程一直持续到A 位点遇到终止密码子(UAG、UGA 或UAA),一旦终止密码子进入核糖体的A 位点,释放因子则被招募到A 位点,多肽链和tRNA 随即被释放,核糖体解离成亚单位,循环用于下一轮翻译[17]。真核细胞内mRNA 翻译机制如图1所示。
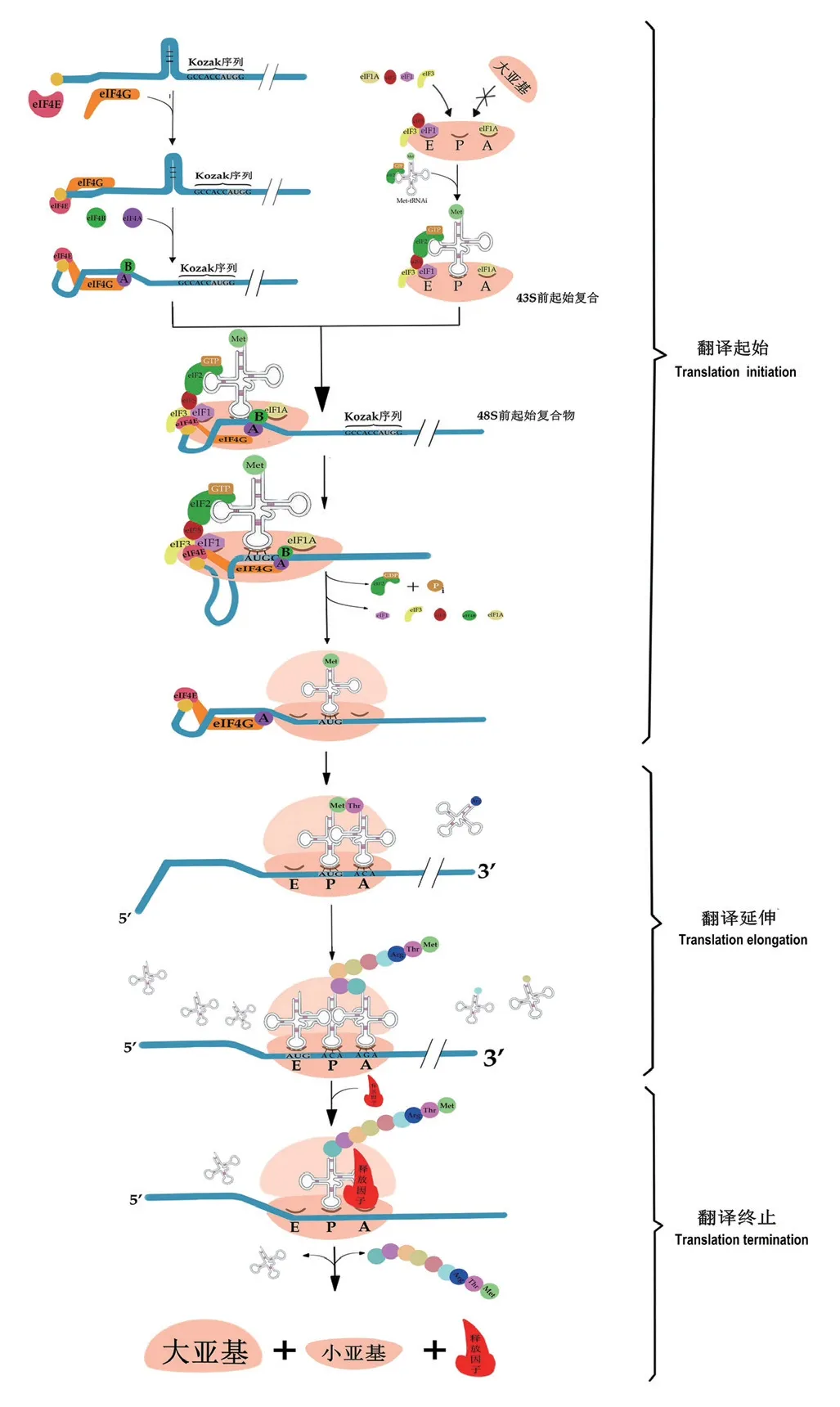
图1 真核细胞内mRNA 翻译机制Fig.1 mRNA translation mechanism in eukaryotic cells
1.2 原核生物mRNA翻译机制
原核细胞内mRNA 的5′ 端通常有SD 序列(Shine-Dalgarno sequence),该序列可与核糖体30S小亚基结合,进而促进mRNA 正确翻译。此外有研究发现,原核细胞内也存在一类可以进行翻译且不含SD 序列的mRNA,但其翻译起始速率低,且具体机制还不清晰。因此,本文以含有SD 序列的mRNA为例进行简要介绍。
在翻译起始阶段,翻译起始因子IF3、IF1 与IF2 的复合物质分别占据30S 小亚基的E、A 位点,其不仅促进翻译起始tRNA 正确的结合,也阻止核糖体大、小亚基的结合[18-19]。随着3 个起始因子的加入,核糖体小亚基已准备好与mRNA、起始tRNA结合。mRNA 的SD 序列与核糖体小亚基形成稳定的SD:aSD 碱基对,促使小亚基与mRNA 的结合;起始tRNA 则在IF2 的招募下,准确地结合于小亚基的P 位点[20-21]。此时,mRNA、核糖体小亚基、起始tRNA、IF3、IF1 以及IF2 共同构成30S 起始复合物。随着起始tRNA 的结合,核糖体小亚基的构象发生变化,IF3 从E 位点脱落[16,22],促使核糖体50S 大亚基与30S 起始复合物结合。IF2 是大亚基初始对接位点,与大亚基相互作用后可激活IF2-GTP 酶的活性,使IF2-GDP、IF1 被释放,最终形成完整的70S起始复合物。此时,mRNA 翻译起始结束,翻译延伸开始。
原核细胞内mRNA 的翻译延伸、终止机制与真核细胞高度相似,都是将携有氨基酸的tRNA 进行易位实现翻译的延伸,通过核糖体识别终止密码子并招募释放因子实现翻译的终止,因此不再做详细介绍,原核细胞内mRNA 翻译机制如图2所示。
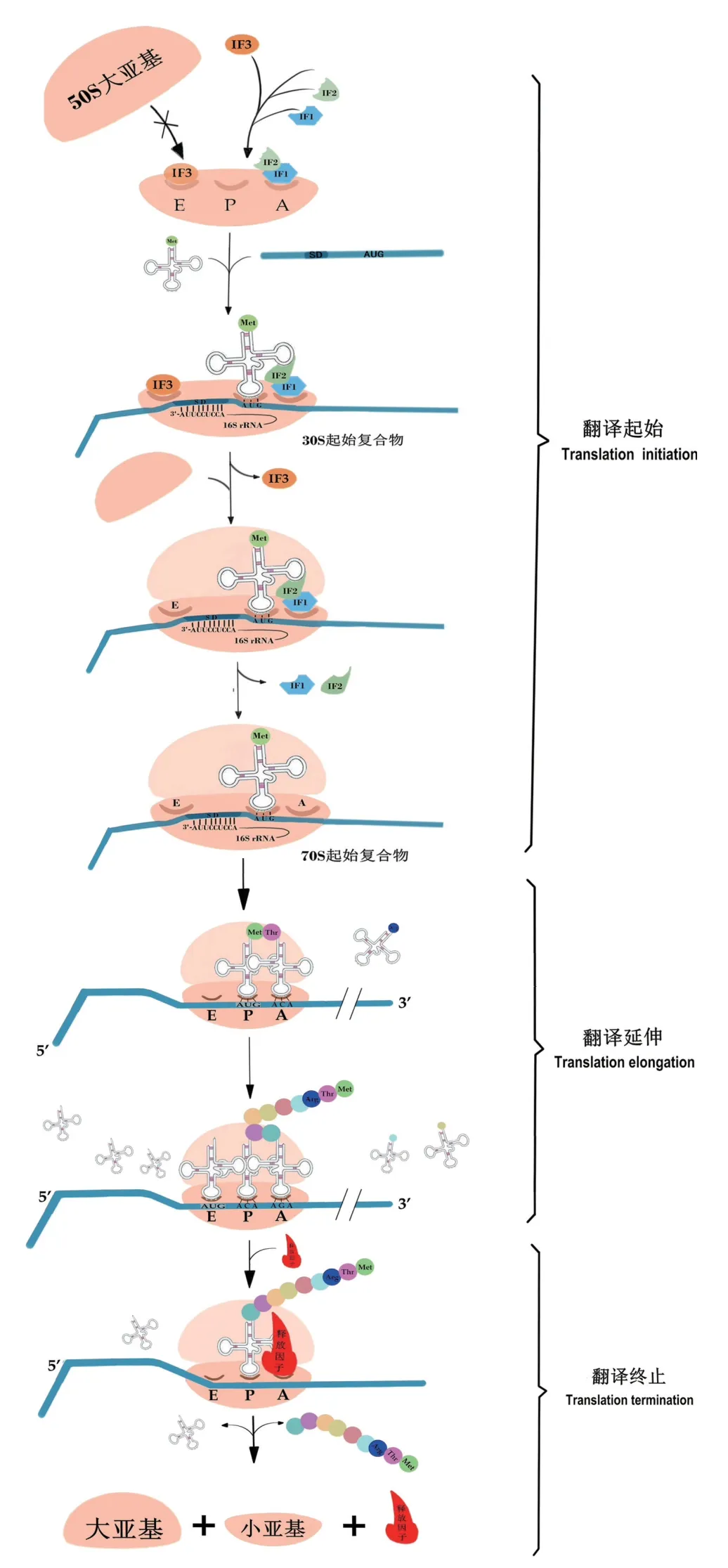
图2 原核细胞内mRNA 翻译机制Fig.2 mRNA translation mechanism in prokaryotic cells
2 翻译速率调控
蛋白质是生命活动的实际执行者,其丰度主要由合成和降解两方面决定[23]。研究发现,蛋白质的降解常数与蛋白质丰度的R2仅为0.10[23],表明蛋白质的降解对蛋白质丰度的影响很小[24],即蛋白质的合成是蛋白质丰度的决定因素。翻译速率不仅是决定蛋白质丰度的关键,也是决定蛋白质折叠的关键因素之一,异常的翻译速率将导致癌症、神经退行等多种疾病[25-26]。关于核糖体翻译速率参数的定义,目前国际尚无定论。2009年Ingolia 等[27]将核糖体的标准化read 数除以mRNA 的标准化read 数,定义为翻译效率。然而mRNA 的翻译速率与其结合核糖体的数量并不总呈正相关,当翻译至mRNA的某一位置停止或暂停时,会导致核糖体聚集,若采用平均核糖体密度描述翻译速率,将会高估这类基因的翻译速率。同时大量研究表明,翻译起始是mRNA 翻译的主要限速步骤,其速率是决定细胞蛋白质水平的关键[28]。在翻译起始阶段,除翻译起始因子影响着翻译的进行,还有mRNA 的“特殊序列”(真核细胞Kozak 序列、原核细胞SD 序列)、mRNA的二级结构和起始密码子等因素决定着翻译的起始速率。
2.1 存在于mRNA的“特殊序列”
2.1.1 真核细胞mRNA 的Kozak 序列 真核细胞mRNA 常以单顺反子的形式存在,其链内的5′帽子结构或IRES 可以被核糖体小亚基识别并结合,进而识别起始密码子、开始翻译。起始密码子AUG之所以可以被核糖体小亚基识别,是因其两侧翼序列(通常为GCCACCAUGG)可以与翻译起始因子结合,进而介导5′帽子结构的mRNA 翻译,这段序列称为Kozak 序列。Kozak 序列组成并不是完全一致的,但AUG 上游-3 位的A 或G,下游+4 位的G 却是高度保守的;同时当序列为GCCACCAUGG、GCCAUGAUGG 时,mRNA 翻译效率最高。有大量研究证明,Kozak 序列是影响起始密码子识别的主要因素之一[29]。例如,酿酒酵母转录本中Kozak 序列是小核糖体亚单位稳定结合位点,其可促进翻译更快的启动[30];在Kozak 序列-6 位的G 以及-1、-2位的C 可以显著地提高mRNA 翻译起始效率[30-31];当上游开放阅读框(upstream open reading frames,uORF)的AUG 起始密码子被放置在一个较不理想的Kozak 序列时,uORF 编码的肽段表达水平较低[32]。
2.1.2 原核细胞mRNA 的SD 序列 1974年John Shine 和Lynn Dalgarno 在大肠杆菌RBS 序列中发现有一段富含嘌呤的序列,其可促进翻译的起始[33],该序列被命名为Shine Dalgarno(SD)。SD 通常指存在于细菌和古细菌等原核生物中的位于翻译起始密码子AUG 上游约8-10 个碱基的核糖体结合位点序列,但也被发现存在于叶绿体和线粒体转录本[34]。SD 序列的组成并不完全相同,其可以与核糖体16S rRNA 的anti-SD 序列(aSD)形成稳定的SD:aSD碱基对,进而促进核糖体的招募和mRNA 的翻译。不同碱基组成的SD 序列与anti-SD 序列的配对程度不同,因此翻译起始效率也有所不同。例如,蓝球藻和莱茵衣藻psbD 基因的SD 序列或起始密码子中引入突变会大大降低翻译效率[35];Barrick 等[36]研究了4 种碱基随机分布的185 种不同组成的SD 序列,以β-半乳糖苷酶为目标蛋白在大肠杆菌中进行表达,其表达量最高和最低相差3 000 多倍。以上表明,SD 序列通过SD:aSD 碱基配对促进翻译起始,且结合程度越强翻译起始速率越高,但其并不是决定翻译的必要条件。此外,SD 序列与起始密码子的相对位置也影响着翻译起始效率。例如,枯草芽孢杆菌的胞内蛋白的SD 序列与起始密码子间隔7-9 个核苷酸的翻译起始效率是间隔4 个核苷酸翻译效率的27 倍[37];RelA 是一类核糖体依赖性合成酶,其通过与SD 序列中的GGAG 序列结合,干扰核糖体与mRNA 的结合,进而抑制翻译起始,但其抑制程度取决于GGAG 相对于AUG 的距离,缩短GGAG 与AUG 之间的距离则会消除RelA 介导的抑制作用[38](图3)。因此,改变SD 序列的碱基组成或改变SD 序列与起始密码子的相对位置都对mRNA翻译起始速率有着重要影响。

图3 ReIA 在SD 区介导的翻译起始抑制Fig.3 Inhibition of translation initiation mediated by ReIA in SD region
2.2 mRNA二级结构
在翻译起始过程中,虽然翻译起始因子可以消除mRNA 的二级结构[33],但对于结构稳定的二级或高级结构却很难消除。因此,mRNA 二级结构对于翻译速率的影响占主导地位,其通过影响核糖体与mRNA 的结合能力阻碍翻译起始[28]。5′非编码区(untranslated region,UTR)是指mRNA 的5′末端帽子结构到起始密码子之间的一段序列,是核糖体识别重要元件之一[39]。该区域通常存在核糖体开关、miRNA 靶位点等可以调控mRNA 二级结构稳定性的调控元件。例如,在枯草芽孢杆菌中xpt-pbuX mRNA 的5′UTR 存在一个对鸟嘌呤高度敏感的核糖体开关,当鸟嘌呤浓度升高时,适体结构域(aptamer domain,AD)结合鸟嘌呤后构象发生变化,表达结构域(expression domain,EPD)的抗隔离子结构消失,导致隔离子茎环结构的形成,使SD 序列和翻译起始密码子 AUG 参与配对,阻止核糖体的结合,抑制 xpt-pbuX 的翻译[40](图4);Gu 等[41]系统地对动物和植物中5′UTR 的二级结构的研究中发现,被miRNA 靶向的mRNA 的5′UTR 往往具有更加稳定的局部二级结构,这种趋势往往存在于动物中,植物中并不存在。此外有研究表明,miRNA 与靶向的mRNA结合后可招募Ago2蛋白沉积于mRNA的靶标;由于Ago2 蛋白的中心结构域与翻译起始因子eIF4E极为相似,因此被沉积于mRNA 靶标上的Ago2 蛋白与m7G 帽子结合,阻止eIF4E 的招募,进而阻止eIF4F 的形成,抑制翻译的起始[42](图5)。此外有研究发现,mRNA 在同一密码子偏好性的前提下,其二级结构的改变可提高mRNA 的翻译效率,而当二级结构不发生改变时,仅使用偏好密码子并不能提高蛋白质的表达,表明稀有密码子的使用是通过弱化mRNA 的二级结构进而促进其翻译[43]。以上结果表明,5′UTR 区内mRNA 的二级结构影响着翻译起始速率,且稀有密码子对翻译速率的影响也是通过强化/弱化mRNA 的二级结构而实现。
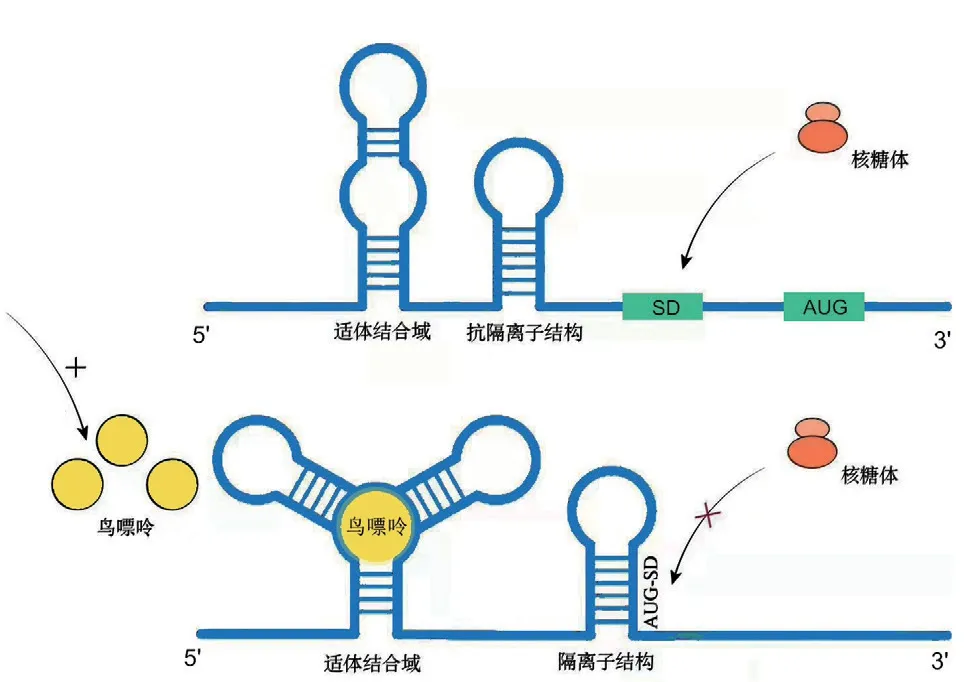
图4 核糖体开关对xpt-pbuX 的调控Fig.4 Regulation of xpt-pbuX by ribosomal switch

图5 Ago2 蛋白对翻译起始的抑制机制Fig.5 Mechanism of Ago2 protein on the inhibition of translation initiation
2.3 起始密码子
细胞内蛋白质的合成并不是从RNA 分子的任何碱基起始的,而是由存在特异序列的AUG 作为起始密码子,开始多肽链的合成。如果没有特异序列的存在,这个密码子则会被单纯地读作链内甲硫氨酸的密码子。Ingoli 等[44]通过对小鼠5 647 个典型蛋白质的起始密码子研究发现,有79%蛋白质的起始密码子是AUG,剩余的21%是同源起始密码子;CUG、GUG、ACG 和UUG 是最常用的同源密码子,而密码子AUU、AUC、AUA 和AGG 出现频率则较少[45]。有研究表明,起始密码子及其后一位密码子类型的改变都影响着mRNA 的翻译起始速率。例如,在真核细胞中,与同源起始密码子相比,翻译起始因子在AUG 处持有更有效的翻译起始[32];Stenstrom[46]在大肠杆菌中利用缺少明显SD 序列特征的基因中发现改变起始密码子下一位的密码子可以促使基因翻译量增加20 倍之多,证明基因表达量与蛋白序列第二位的密码子有关,并且高表达蛋白的第二位氨基酸倾向使用腺嘌呤含量高的密码子编码。此外,5′UTR 区的非AUG 起始的uORF 通常对下游基因的翻译产生抑制作用;且该区的AUG 密码子也会干扰核糖体的招募,从而导致翻译起始效率降低[30]。
3 翻译研究技术
除探究mRNA“特殊序列”、mRNA 的二级结构和起始密码子对翻译起始速率的影响外,整体评估mRNA 翻译速率对于蛋白质的表达有着重要的意义。mRNA 测序技术非常完善,通过高通量测序技术探究处于翻译状态的mRNA,既可以间接反应蛋白质的翻译水平,又可以检测到基因的可变剪接甚至发现新蛋白质分子[1-2]。因此,利用高通量测序技术对正在翻译的mRNA 测序和分析成为当今研究的热点。翻译组研究最为常见的是多聚核糖体图谱技术(polysome profiling),此外也有最近发展起来的翻译谱分析技术(RNC-RNA-Seq)、核糖体图谱技术(ribosome profiling)、核糖体亲和纯化技术(ribosome affinity purification,RAP)。翻译组研究技术比较见表1。

表1 翻译组研究技术比较Table 1 Comparison of techniques in translatome
3.1 多聚核糖体图谱技术
核糖体是细胞内部密度最大的生物分子,单条mRNA 可以同时与多个核糖体结合形成分子量更大的多聚复合物。利用抗生素放线菌酮可将正在翻译的RNA 固定,阻止核糖体内tRNA 释放[47],进而阻止翻译延伸。随后利用蔗糖密度梯度离心法将分布在不同梯度溶液中游离的RNA、核糖体大/小亚基以及结合有不同数目核糖体的mRNA 进行分离。根据结合核糖体数目的不同对mRNA 进行分离,并利用Northern 杂交、RT-qPCR[48-49]和微阵列分析[50-53]等方法对每组收集到的特定mRNA 进行分析(图6)。此外,若对正在翻译的RNA 进行整体分析,可以通过构建RNA-Seq 文库、高通量测序[54-56]实现。多聚核糖体图谱技术在人类癌症的治疗、非编码RNA 编码小肽的发现及植物在逆境胁迫的应答等多方面都得到广泛应用。例如,Polenkowski 等[57]通过多聚核糖体图谱技术发现在肝癌细胞(HCC)中表达且可以编码功能性小蛋白C20orf204-189AA 的Linc00176,其可能是HCC的特异性靶分子,且C20orf204-189AA 小蛋白也可以增强核糖体RNA 的转录,进一步促进HCC 的增殖;高温胁迫是全球小麦生产的主要限制因素,通过CRISPR /Cas9 的基因编辑和多聚核糖体图谱等技术分析发现基因TaMBF1c显著影响着热响应因子的翻译效率,且与热休克蛋白的翻译密切相关,其结果揭示了TaMBF1c通过翻译调控小麦的热应激反应[58]。
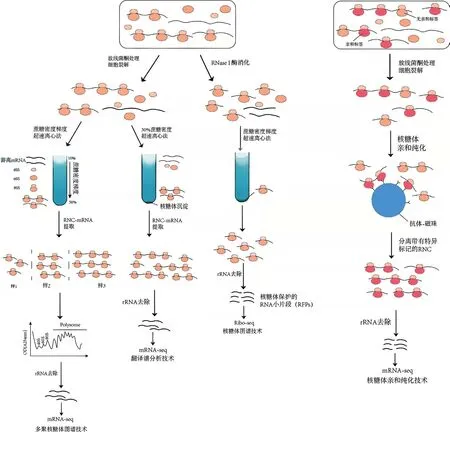
图6 翻译组研究技术流程Fig.6 Technical process of translationomics
多聚核糖体图谱技术能够获取正在翻译的mRNA 的全长信息,但没有办法获知mRNA 内部的翻译信息,例如核糖体位置、阅读框位置、翻译暂停区、uORFs 等。该实验相对容易、适用范围广,但实验操作过程复杂、样本需求量大[56]。此外,蔗糖密度离心的体积通常较大[27],且在高浓度的蔗糖溶液中常掺有脂筏、假多聚核糖体等高分子量复合物[59],这些都大大增加了获取核糖体-新生肽链复合物(ribosome nascent-chain complex,RNC)中的mRNA 即翻译成蛋白质的mRNA 的难度,进一步局限该技术的发展。
在评估细胞内翻译活跃程度方面,多聚核糖体图谱可根据mRNA 结合核糖体数目判断该mRNA 翻译的活跃程度,且根据mRNA 在高浓度和低浓度蔗糖溶液的比例、正在翻译的mRNA 与总mRNA 之间的比例共同判断细胞的活跃程度。
3.2 翻译谱分析技术
有研究发现,若一条mRNA 结合过多的核糖体则易发生核糖体“堆积”,阻碍翻译延伸,降低翻译速率。然而,部分mRNA 虽只结合单个核糖体,但其翻译状态却异常的活跃。如在公认的翻译活跃的HEK293 细胞和处于富营养培养基上对数生长的大肠杆菌中,其单核糖体组分占有主要地位[60-61]。将单核糖体单体分离并放置于无细胞翻译体系中,其核糖体可以继续翻译生成蛋白质[61]。这都说明翻译的活跃程度与mRNA 上核糖体的数量并不总呈正相关,但只要mRNA 与核糖体结合即可表明其处于翻译状态[62]。基于该理论,2013年张弓实验室开发了RNC-RNA-Seq[3],与多聚核糖体图谱技术类似,利用超速离心的方法将正在翻译的mRNA 与游离的mRNA 分离并对翻译中的mRNA 测序建库,不同的是RNC-RNA-Seq 是采用单一浓度蔗糖溶液(30%蔗糖溶液)将正在翻译的mRNA 与游离的mRNA 分离。该技术通常与RNA-Seq 技术相结合,共同探究动植物及细菌等在体内的转录和翻译调控。例如,Luo 等[63]发现甜菜碱是通过调节基因的翻译过程发挥降脂作用,其中Gpc1是降脂的关键基因;Zhang等[64]在胶质母细胞中发现LINC-PINT 可编码多肽,且该肽可直接与聚合酶相关因子复合物(PAF1c)相互作用,抑制多种癌基因的翻译过程,进而抑制胶质母肿瘤细胞的增殖。
与多聚核糖体图谱相比,RNC-RNA-Seq 简化了分离RNC-mRNA 的程序,有效地解决了多聚核糖体图谱技术中溶液体积大、RNC-mRNA 浓度低的问题[27]。此外,实验中得到的RNC-mRNA 仍保留翻译活性,给予合适的缓冲液即可恢复翻译能力[61]。不足的是RNC-RNA-Seq 依旧没有办法获取mRNA内部翻译信息,且单一浓度的蔗糖溶液使得沉淀物中不仅包含脂筏、假多聚核糖体等[59]高分子量复合物,还含有核糖体的大小亚基、单核糖体等,增加了获取mRNA 信息的难度。
由于RNC-RNA-Seq 探究的是细胞内mRNA 分子是否进行翻译,对某个基因或细胞内整体翻译速率的水平还无法探知。但该技术与RNA-Seq 结合可共同分析翻译过程中细胞整体或某个基因的翻译水平。
3.3 核糖体图谱技术
2009年由Weissman 课题组首次发表核糖体图谱技术(Ribo-Seq)[65],其利用核糖核酸酶I(ribonuclease I,RNase I)降解游离和不被核糖体保护的RNA 片段,并通过蔗糖梯度超速离心法获取被核糖体保护的RNA 片段(ribosome footprints,RFPs),随后去除核糖体,并将RNA 片段进行高通量测序和生物信息学分析,最终获取正在翻译的RNA 信息。Ribo-Seq 的精度是单密码子级别,可精准获取某基因内部的翻译信息,如核糖体分布位置、起始密码子位置、程序性框移动、uORF 等[66]。除Ribo-Seq 外,Pelechano 等[67]建立的5PSeq 技术也可以在单碱基分辨率下高通量捕获核糖体的位置信息。因此,上述两种技术在开放阅读框(open reading frames,ORF)的发现、翻译起始位点的发现及翻译暂停等方面有着得天独厚的优势[62,65]。例如,Sotta 等[68]采用Ribo-Seq 确定了高粱全基因组中快速翻译的阅读框,检测到4 843 个主开放阅读框(main ORF,mORF),并发现许多未被注释的翻译事件;Van’t Spijker[69]在人类G4C2基因的上游内含子中发现3 个起始位点,且该基因在核糖体易位过程中出现停顿现象。
虽然Ribo-Seq 有诸多优点,但其存在样本需求量大、成本高且操作要求高等问题[62]。此外,Ribo-Seq 也存在一定的局限性。例如,RNase I 不会降解与核糖体结合且被RNA 结合蛋白保护的或双链的RNA[70],此类RNA 在实验中会被剔除,进而遗漏部分正在翻译的RNA 信息;该技术只分析与核糖体结合的区域,并不能获得与翻译调控相关的非翻译区的信息[27];实验通常只选取25-35 nt 的RFPs,而核糖体保护的RNA 片段长度差异很大(通常为19-75 nt),忽略其他大小的RFPs 片段,易造成翻译信息的缺失。因此我们还需要从不同方面不断优化实验,提出更加合理的实验方案。
通过Ribo-Seq,可以清楚地了解每条RNA 结合核糖体的数目、位置等信息,因此可以根据基因结合核糖体的数目初步判断翻译的活跃程度。此外,也可以结合转录组数据计算整体或某基因的翻译水平。
3.4 核糖体亲和纯化技术
核糖体亲和纯化技术(RAP)最早是由Gerbei课题组于2002年发表[27],与其他翻译组技术不同,利用亲和纯化以及组织特异性启动子可在目的细胞内特异性表达这两大原理,实现对样品中难以分离的目的细胞进行翻译水平的探究。其在核糖体内RPL25p 或RPL16a 蛋白的C 末端连接亲和标签(如His、FLAG 和eGFP 等),这两类蛋白可被组织特异性启动子启动,纯化后获得过表达核糖体蛋白细胞系,随后采用抗体与抗原特异性结合的原理将含有标签的RNC-mRNA 进行分离,最后将被标记的mRNA 进行测序建库分析[24]。该技术在检测组织中特异性表达的mRNA、细胞的分离和建系等方面有着举足轻重的地位。例如,Heiman[71]在中枢神经系统细胞中发现上千个在细胞中特异性表达的mRNA,这些mRNA 在RNA-Seq 中很难被检测到;Corbacho[72]利用RAP 对斑马鱼细胞群中的核糖体进行遗传标记,获得基因在视网膜、骨骼肌、颌骨等组织中特异性表达的细胞系。
RAP 技术不需要对RNC-mRNA 进行广泛的固定、超速离心分离或酶消化,可以在最小体外刺激下保持较完整的翻译中RNA 图谱,而且可以准确的定位在目的细胞,无需考虑来自其他组织、细胞的污染,获得与该细胞表型相关性更高的数据信息。但实验流程繁琐冗长、成本昂贵,且不能获得RNA的阅读框位置、翻译暂停区、uORFs 以及核糖体数量和位置等信息;此外,过表达核糖体蛋白会干扰正常生理状况,携有标签的核糖体的翻译性能也与正常核糖体不同,因而不能准确地反映正常生理状况下的翻译状况[27]。
与RNC-RNA-Seq 相同,都不能获取核糖体数目信息。因此,在计算整体或某基因的翻译速率时都需要与RNA-Seq 数据相结合,共同分析细胞整体或单基因的翻译水平。
4 总结与展望
中心法则阐述了遗传信息从DNA 通过转录为RNA,再经翻译合成蛋白质的基本生物学过程。蛋白质是发挥生物学功能的实际执行者,但其组成多样、构象复杂且结构稳定等都严重阻碍着蛋白质组学的发展。翻译速率是蛋白质丰度的决定性因素,众多学者通过mRNA 结合核糖体的数目判断翻译的快慢。事实上,该方法在一定条件下是准确的;当核糖体在mRNA 上“堆积”时,过多的核糖体则会阻碍mRNA 的翻译,利用该方法评估mRNA 的翻译速率是不准确的。值得注意的是,并非高速的翻译就能获得最佳的表达效果,高速的翻译会使多肽链错误折叠的概率增加,进而形成无活性、不可溶的包涵体。所以翻译速率应当与蛋白质的折叠速率相“匹配”,这其中必然存在着某些重要的调控因子调控着二者的速率,还需进一步的挖掘和发现。当前,从基因组到转录组的调控机理研究得较为清楚,而从转录组到蛋白组的调控机理研究还非常薄弱,而翻译组恰恰是连接转录组和蛋白组的桥梁。因此,通过翻译组探究正在翻译的mRNA 已经成为当前研究的热点。利用该技术可帮助我们清晰地了解细胞内RNA 的翻译状态,有利于发现更多具有编码能力的非编码RNA,这为蛋白数据库和分子生物学理论的补充和完善,对生命科学的发展具有重要的意义。
