直线型稠环芳烃的分子结构及振动光谱的理论研究
2023-01-04靳佳平陈寿勇况迪森赖仕全
靳佳平,陈寿勇,况迪森,岳 莉,赖仕全
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
直线型稠环芳烃又称为并苯,是一类由苯环渺位缩合而成且呈直线排列的多环芳烃(Polycyclic aromatic hydrocarbons,PAHs),具有独特的线型π共轭电子体系,可作为有机半导体材料,在分子电子学和自旋电子学中具有重要的应用前景[1-2]。小型并苯,如萘(并二苯)、蒽(并三苯),可从煤焦油或重质石油馏分中通过精馏分离得到,而环数n≥4的大型并苯在自然界中还未发现,通常需要多步合成反应才能制备[3-5]。但是这些大型并苯的稳定性差,导致合成非常困难,进而对其结构、性质分析不深入。因此,采用理论方法研究它们的分子结构和微观性质显得格外重要。研究这些具有不同苯环数的直线型稠环芳烃的几何结构,还有助于理解煤、沥青、渣油等富芳烃有机物在成焦过程中石墨微晶的增长机制[6]。
密度泛函理论(Density functional theory,DFT)是研究多电子体系电子结构的量子化学方法。近年来,利用DFT研究PAHs分子结构与性质越来越受到重视[7]。邹乔等[8]利用DFT研究菲的分子结构与拉曼光谱。范青杰等[9]采用B3LYP方法研究并四苯的分子结构与振动光谱。杨惠纯等[10]采用电子紧束缚模型计算并五苯分子的电子能级。Yamakita等[11]通过DFT研究低聚并苯(n=2~5和10)的分子振动与声子色散关系。曾娅玲等[12]则采用B3LYP方法对美国EPA优先控制污染物中的16种PAHs的拉曼光谱进行辨识。有关PAHs分子的理论计算研究近年来取得大量成果,但大多数工作都集中在单个分子的结构和特定性质上,而系统研究一系列PAHs分子结构和微观性质的报道极少。
本文利用DFT中的B3LYP方法,在6-311++G(d,p)基组下对渺位增长的2~8环的直线型稠环芳烃萘、蒽、并四苯、并五苯、并六苯、并七苯、并八苯共七种分子进行分子结构、振动光谱、原子电荷、HOMO-LUMO能隙、分子静电势分析等研究。
1 分子结构优化
先构建萘、蒽、并四苯、并五苯、并六苯、并七苯、并八苯的均键型分子模型,再利用密度泛函理论,在B3LYP/6-311++G(d,p)水平下采用Gaussian 09W程序包,对七种分子进行分子频率计算和结构优化。由于各分子的最小频率均为正值且无虚频,表明优化后的结构为稳定构型,而且碳原子间的二面角为180°,也表明这些分子为直线型分子。另外,萘、蒽、并四苯、并五苯、并六苯、并七苯及并八苯的全局最小能分别为-1.050×104、-1.468×104、-1.887×104、-2.305×104、-2.723×104、-3.141×104、-3.559×104eV,表明苯环链越长的分子,要保持其稳定结构则需要更多能量。
图1给出七种分子优化后的几何结构、原子编号以及根据键长、键角计算出的分子长宽尺寸。随环数增多,分子的长度逐步增加,从2环的0.674 6 nm增加到8环的2.148 0 nm,增长了2.18倍,而宽度仅有轻微增加,从2环的0.497 2 nm增加到8环的0.498 9 nm。七种分子的结构均具有明显的对称性,所以其C—C键和C—H键的键长也呈对称分布。表1仅列出各分子中独立的C—C键和C—H键的键长。在这些直线型芳烃分子中,除了萘外,其余分子周环上的C—H键长仅有三种类型(0.108 4 nm、0.108 5 nm和0.108 6 nm),几乎不受分子苯环数增加的影响,且各分子中左右两侧苯环上的C—H键长(Ⅰ、Ⅱ)略低于中间苯环上的C—H键长(Ⅲ)。而C—C键长的分布则复杂得多,随分子中苯环数的增多而增加,2环的萘为4种,3环的蒽为5种,4环的并四苯为6种,5环的并五苯为8种,6环的并六苯为10种,7环的并七苯为11种,8环的并八苯为13种。
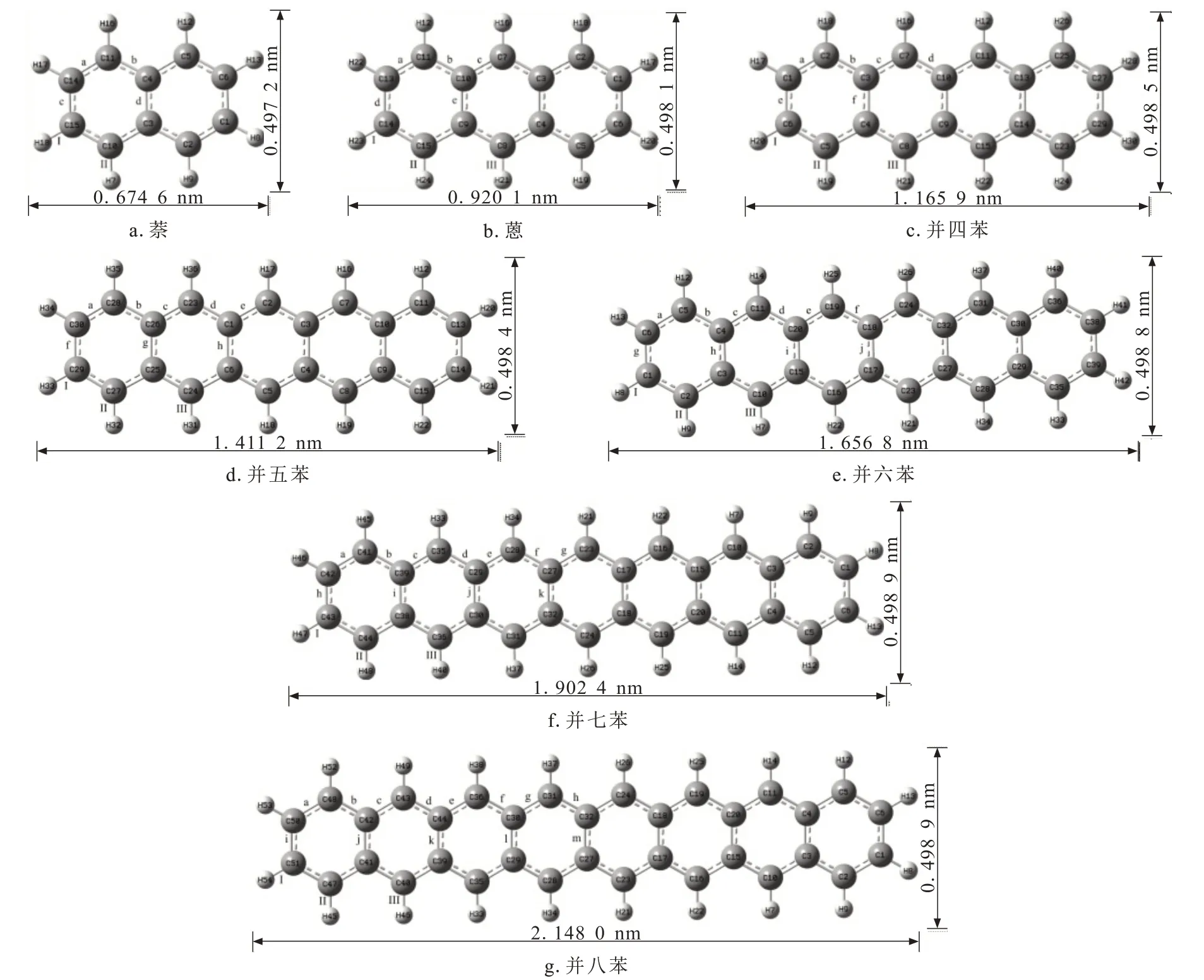
图1 七种分子优化结构的几何参数Fig.1 Geometric parameters of optimized structure for seven molecules

表1 七种分子的键长分布,nmTab.1 Bond length distributions of seven molecules,nm
2 振动光谱
2.1 振动模式
对优化后的分子结构进行频率计算,得到各分子详细的振动模式信息。七种多环芳烃的振动模式主要包括苯环骨架变形、C—C伸缩、C—H摇摆、C—H伸缩以及各种形式的耦合[12]。萘共有48种振动模式,包括33种面内对称振动、15种面外对称振动;蒽共有66种振动模式,包括45种面内对称振动、21种面外对称振动;并四苯共有84种振动模式,包括57种面内对称振动、27种面外对称振动;并五苯共有102种振动模式,包括69种面内对称振动、33种面外对称振动;并六苯共有120种振动模式,包括75种面内对称振动、45种面外对称振动;并七苯共有138种振动模式,包括88种面内对称振动、50种面外对称振动;并八苯共有156种振动模式,包括105种面内对称振动、51种面外对称振动。其中,萘有21种红外活性振动、28种拉曼活性振动;蒽有30种红外活性振动、36种拉曼活性振动;并四苯有46种红外活性振动、64种拉曼活性振动;并五苯有64种红外活性振动、83种拉曼活性振动;并六苯有68种红外活性振动、83种拉曼活性振动;并七苯有70种红外活性振动、94种拉曼活性振动;并八苯有79种红外活性振动、104种拉曼活性振动。随着分子苯环数的增加,振动模式明显增多,振动形式也越复杂。
2.2 红外和拉曼光谱
七种分子的计算红外光谱和拉曼光谱如图2所示,与SDSB图谱数据库中实测图谱的出峰数目和位置一致。振动频率主要分布在3 000~3 300、1 000~1 800、700~1 000、0~700 cm-1四个区。在3 000~3 300 cm-1区,七种分子在3 050 cm-1左右均有一个明显的强峰,并且并四苯、并五苯、并六苯、并七苯、并八苯中在3 030 cm-1左右又分裂出一个强肩峰,这些峰都归属于芳环中不同类型的C—H伸缩振动。在1 000~1 800 cm-1区,振动情况较为复杂,存在多种振动重叠现象,但大多数振动源于苯环的C—C伸缩振动,同时伴随着C—H面内弯曲振动及苯环骨架振动或变形。该区域红外峰相对较弱,而拉曼峰信号则非常强,表明拉曼光谱更适合表征稠环芳烃的碳骨架振动。在700~1 000 cm-1区,红外吸收峰则很强,而拉曼光谱在此区域内则几乎无振动信号,此区域峰归属于不同类型的C—H面外弯曲振动,主要包括1H(880~900 cm-1)和4H(720~750 cm-1)两种振动模式[13]。在0~700 cm-1区,两种光谱都仅有零星的峰,且强度都较弱,主要归属于苯环骨架的不同类型面外振动,如:碟形振动、折叠振动以及扭转振动等[14]。

图2 七种分子的计算红外光谱和拉曼光谱Fig.2 Calculated infrared spectra and Raman spectra of seven molecules
3 分子电荷和电势分析
3.1 Mulliken电荷分布
七种分子中C、H原子的Mulliken电荷分布详见表2。随分子中苯环数增加,C、H原子电荷分布越复杂,类型越多。七种分子结构具有对称性,所以其原子电荷也呈对称分布。通常,居于分子左右两侧的C原子具有最大的负电荷,而靠近中心的C原子的电负性减弱,中心C原子具有最大正电荷。随分子苯环数增加,C原子的电荷有减小的趋势。在七种分子中,C原子电荷既有正也有负,有规律性的交替出现,表明它们均形成了共轭碳网体系。

表2 七种分子的Mulliken原子电荷分布Tab.2 Mulliken atomic charge distributions of seven molecules
3.2 分子静电势
分子静电势(Molecular electrostatic potential,MEP)是分子的一个重要微观性质参数,用于研究分子间的相互作用、反应位点以及分子识别[15]。本文基于自洽场(Self-consistent field,SCF)理论,利用总电子密度对优化后的分子结构作图,得到分子静电势的3D图,如图3所示。深色代表负电势区域,浅色代表正电势区域,二者之间为过渡区。七种分子的碳网上方均形成明显的负电势区,而其四周的H原子则形成正电势区。苯环数越多的分子,负电势区域越浅,表明其形成更大的离域π键,结构更稳定,亲电活性变弱,不易发生亲电取代反应。

图3 七种分子静电势的3D图Fig.3 3D images of electrostatic potential for seven molecules
3.3 前线分子轨道
前线分子轨道作为量子化学的一个重要参数,对分子理化性质的研究十分重要,包括最高占据分子轨道(Highest occupied molecular orbital,HOMO)和最低未占据分子轨道(Lowest unoccupied molecular orbital,LUMO)。HOMO能(EHOMO)越小,表示其失去电子的能力越强,而LUMO能(ELUMO)越小,则表示其接受电子的能力越弱。二者之差称为HUMO-LUMO能隙(ΔE),是电子从HOMO跃迁到LUMO所需的最低能量,可反映物质导电性、发光性的强弱。
利用GaussView 6.0程序对优化后的分子结构进行分子轨道分析,得到七种分子的EHOMO、ELUMO和ΔE,结果列于表3中。从萘到并八苯,EHOMO越来越大,而ELUMO越来越小,表明随苯环数增加,分子失去电子能力和接受电子能力均逐步减弱。这也反映了苯环数增加,分子的反应性减弱,稳定性逐渐变好,这与MEP分析结果一致。能隙ΔE也随苯环数增加而逐步减小,从萘的4.748 eV减小至并八苯的1.230 eV,表明大型并苯更易受外场激发,更适合作为导电或发光材料。
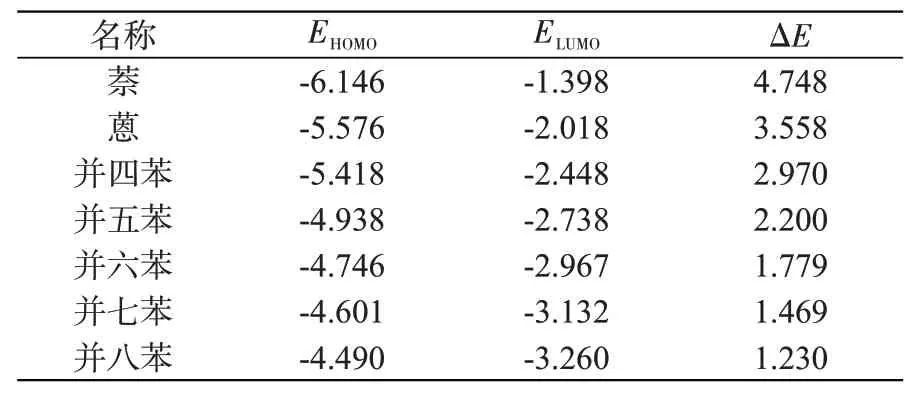
表3 七种分子的HOMO、LUMO能和能隙,eVTab.3 HOMO,LUMO energies and energy gaps of seven molecules,eV
4 分子性质参数分析
4.1 极化率
极化率(α)是指分子在外场下产生极化的程度,是与分子光学性质密切相关的重要参数。对分子结构优化后,根据在x、y、z方向的精确极化率(αxx、αyy、αzz),计算极化率(-α)

七种分子的极化率如表4所示。七种分子的极化率都较大,这主要归因于它们都有离域的π电子结构,而且含苯环数越多的分子,其极化率越大。αxx、αyy、αzz均随分子苯环数增多而增大,各分子在x、y、z方向的极化率的大小顺序为:αxx>αyy>αzz,表明这些分子光电各向异性,且在大型并苯中这种现象越发明显。

表4 七种分子的极化率Tab.4 Polarizabilities of seven molecules
4.2 能量及热力学参数
七种分子的零点振动能EZPV,以及在平动、转动、振动时的能量E、恒容热容C、熵S等性质参数详见表5。零点振动能、总动能(Etotal)、总热容(Ctotal)和总熵(Stotal)均随分子苯环数增多而增大。分子每增加一个苯环,其零点振动能、总动能分别增加120~130 kJ/mol,总热容、总熵则分别增加50 J/(mol⋅K)左右。在动能中,所有分子的平动能(Etranslational)均等于转动能(Erotational),但二者远低于振动能(Evibrational)。在恒容热容中,振动的贡献占主导地位,平动和转动的贡献则相同。三种运动模式对分子熵的贡献比较复杂,在小型并苯(萘、蒽)中,平动的贡献最大,转动次之,振动最小;而在大型并苯(n≥4)中,随分子环数增加,振动的贡献逐渐变为最大,如在并八苯中,振动熵占总熵的50%左右。
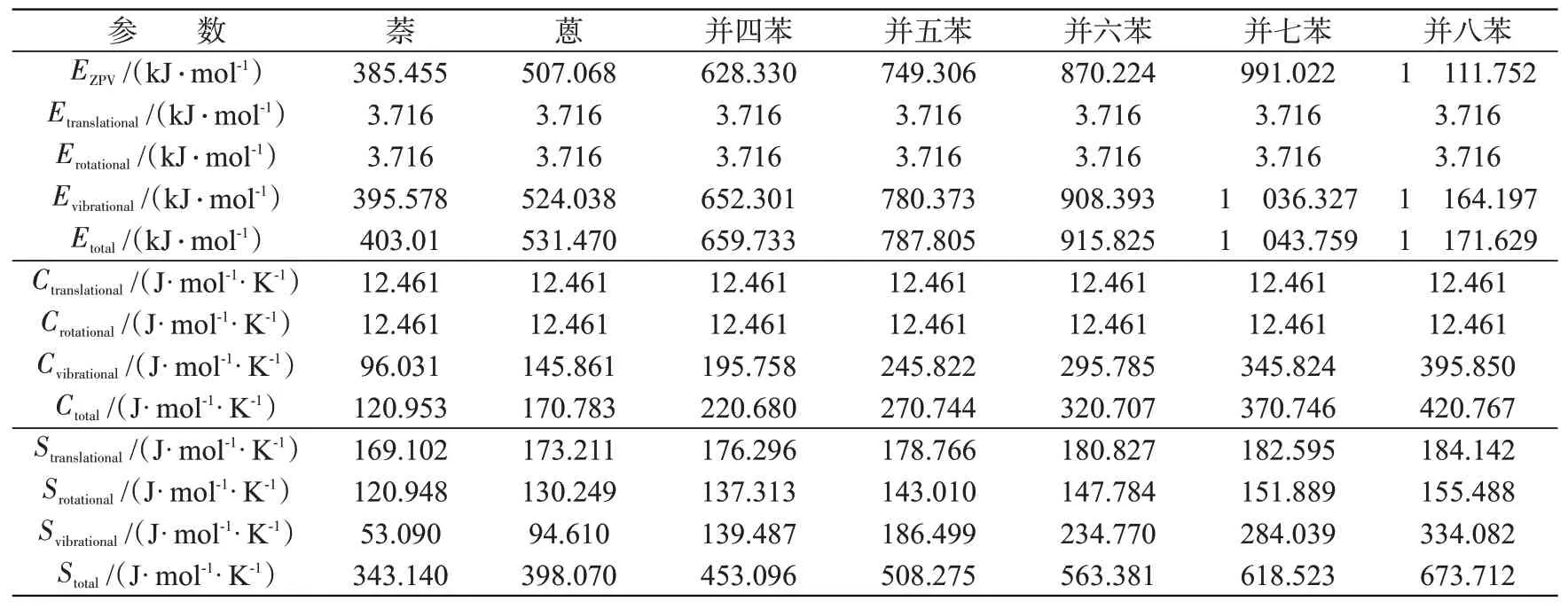
表5 七种分子的各类能量及热力学参数Tab.5 Various energies and thermodynamic parameters of seven molecules
5 结论
利用密度泛函理论对萘、蒽、并四苯、并五苯、并六苯、并七苯、并八苯七种直线型多环芳烃进行结构优化和频率计算,得到其结构参数、振动光谱及微观性质数据。分子中苯环数越多,其C—C键的键长种类则越多,但C—H键的键长主要在0.108 4~0.108 5 nm范围。七种分子的红外活性振动峰主要分布在3 000~3 300、700~1 000 cm-1两个区,而拉曼活性振动峰主要在1 000~1 800 cm-1区。理论计算频率与实测频率一致。周环H中,左右两侧的H具有更高的反应活性,易被取代。随分子中苯环数的增加,HOMO-LUMO能隙从萘的4.748 eV减小至并八苯的1.230 eV,而极化率从萘的116.647 a.u.增大至并八苯的642.471 a.u.,这些物质在光电材料方面有良好的应用前景。
