生酮饮食对基因变异相关癫痫疗效分析
2022-12-28胡笑月井淼王艳萍华颖
胡笑月 井淼 王艳萍 华颖
无锡市儿童医院神经内科(江苏无锡 214023)
生酮饮食(ketogenic diet therapy,KDT)是一种高脂肪、低碳水化合物的特殊饮食疗法,从20世纪初就开始应用于药物难治性癫痫患儿。目前KDT已被证实在多种癫痫综合征中疗效显著[1-3],并成为儿童难治性癫痫的重要治疗方法。然而,KDT的疗效也常因人而异,因其对饮食方案的限制性和潜在的不良反应,对患儿和其监护人而言仍是一个不小的挑战。因此,能预测KDT的疗效将有助于筛选出最合适的患儿,以达到医疗资源的最优化使用。随着二代测序技术的发展,越来越多的难治性癫痫的致病基因得以发现,也有学者开始致力于探索癫痫的精准治疗。2018年,韩国学者回顾性分析了73例检测出有致病基因的癫痫患儿,发现SCN2A、KCNQ2、STXBP1、SCN1A基因变异的患儿对KDT尤为有效[4]。总结不同基因型与KDT疗效的关系,可以使更多有可能受益的患儿尽早启动KDT。现对10例基因变异相关癫痫患儿的KDT疗效进行回顾性分析。
1 资料与方法
1.1 研究对象
2017年4月至2021年3月无锡市儿童医院收治的KDT 治疗的10 例基因变异相关癫痫患儿(符合2017年国际抗癫痫联盟诊断标准)。纳入标准:①有明确的癫痫发作;②接受二代测序基因检测,筛选出了相关的致病基因;③启动了经典KDT,且治疗过程依从性好。排除标准:①排除大脑结构异常、颅内感染、自身免疫性脑炎等其他引起癫痫的病因;②失访患者。本研究获无锡市儿童医院伦理委员会的批准,患儿家长知情同意。
1.2 临床资料及辅助检查
回顾性收集10例患儿的临床资料,包括性别、年龄、围生期病史、生长发育史、家族史、用药史、临床表现、治疗过程、不良反应等。所有患儿在KDT前均行血生化、头颅磁共振成像(MRI)、血尿代谢筛查等检查。
1.3 基因检测
委托康旭医学检验所、凯昂医学检验实验室进行检测,采用目的基因靶向获取二代测序或全外显子测序分析。
1.4 随访及疗效评级
分别在KDT启动后1、3、6和12个月进行随访,随访内容包括KDT持续时间、癫痫控制及发育改善情况。最后1次随访时间为2021年9月。疗效评价:KDT 治疗前3 个月平均每月的发作频率作为基线,与基线比较,将癫痫发作控制情况分为3 种:无发作、发作减少≥50%、发作减少<50%,将发作减少≥50%认为有效,发作减少<50%或增加为无效。
2 结果
2.1 一般资料
10例患儿中男4例,女6例;发病年龄3天~1岁4个月,平均为5.5个月(表1)。所有患儿均存在发育落后情况。患儿出生史均正常,均无热性惊厥或癫痫家族史。血生化、血尿代谢筛查、头颅MRI等均未见异常。
2.2 基因检测及临床表现
10例患儿均检测到致病基因,其中9例(例1~例9)为新生变异,1 例(例10)男性患儿为遗传自健康母亲的半合子变异。2例为SLC2A1基因变异所致的葡萄糖转运子1缺陷综合征(glucose transporter type 1 deficiency syndrome,GLUT1-DS),脑脊液检查糖水平均低于2.2 mmol/L,脑脊液糖/血糖指数<0.45,分别为错义变异(c.476T>C,p.Leu159Pro)和移码变异(c.164_165delinsTTCA,p.Ser55AsnfsTer24);5例为SCN1A基因变异引起的Dravet综合征,2例为错义变异(c.5606T>C,p.Phe1869Ser、c.580G>A,p.Asp 194 Asn),1 例无义变异(c.3733 C>T,p.Arg1245Ter),1例移码变异(c.135delC)及1例剪切变异(c.3705+2 T>C);1 例为KIF 1 A基因变异(c.296C>T,p.Thr99Met)所致的PHEO(progressive encephalopathy with edema,hypsarrhythmia and optic atrophy syndrome)样综合征,具有肌张力高、肌无力、脑电图高度失律、婴儿期逐渐出现小头畸形等特征,病初表现为局灶性发作和痉挛发作,后期出现肌阵挛发作,4月龄时行头颅MRI检查未见脑萎缩;1例为SCN2A基因错义变异(c.468G>C,p.Lys156Asn)引起的发育性癫痫性脑病(Developmental and Epileptic Encephalopathy,DEE)患儿,生后3天起即出现抽搐发作,具有丛集性发作的特点,最多时每日发作高达几十次;1例为PIGA基因变异(c.241C>T,p.Arg81Cys)引起的多发先天畸形-肌张力低下-癫痫综合征(multiple congenital malformation-hypotoniaepilepsy syndrome type 2,MCAHS),患儿表现为发育迟缓、肌张力低下、癫痫、高腭弓及面色发黄。见表1。

表1 10例患儿的临床特征及基因信息
2.3 生酮治疗疗效
治疗前所有患儿均应用≥2 种抗癫痫药物而癫痫控制欠佳,其中发作频繁(癫痫发作每月至少4次以上)的9例患儿启用KDT后,4例在1个月内起效,其中2例在1周内即达到癫痫无发作。随访至2021年9月,有效的患儿为7例,其中无发作4例。4例无发作患儿的变异基因为SLC2A1基因2例,SCN2A基因1例,PIGA基因1例;3例SCN1A基因变异的患儿癫痫发作减少≥50%,其中1例(例4)后续添加吡仑帕奈治疗目前已有1年无发作。2例SCN1A基因患儿发作减少<50%,1例也已添加吡仑帕奈治疗1月,疗效待观察。KIF1A基因变异的患儿KDT治疗后癫痫发作无明显改善,后应用ACTH治疗仍有间断发作(表2)。KDT期间,10例患儿中有7例出现轻微的短期不良反应(表2),5例存在腹泻、纳差及便秘等胃肠道症状,予添加益生菌治疗后好转。1例出现肾结晶,予增加饮水后缓解,1例存在骨密度降低,予补充维生素D和钙剂后好转。
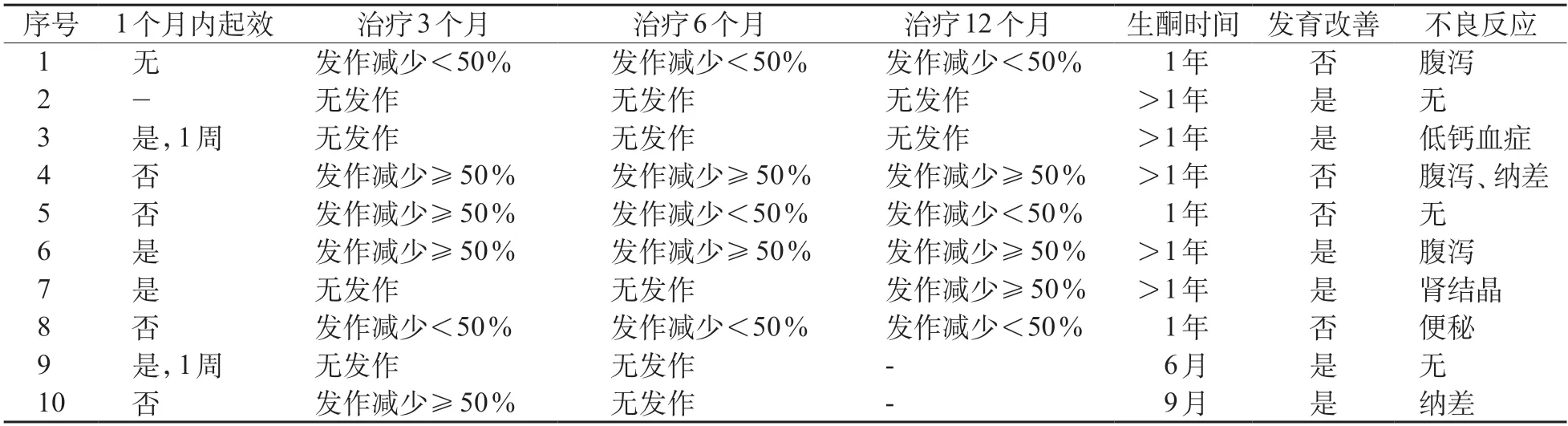
表2 10例患儿KDT随访情况
3 讨论
KDT代谢过程中会产生的大量酮体,包括乙酰乙酸、β-羟基丁酸和丙酮,被认为具有抗癫痫作用。KDT治疗癫痫的主要机制包括:①增加抑制性神经递质GABA的合成,或减少兴奋性神经递质谷氨酸的合成;②激活兴奋性神经元腺苷A1受体,起到抗惊厥作用;③增强线粒体的合成和能量代谢;④抑制N型电压门控钙通道,使谷氨酸释放减少等[5]。
葡萄糖转运体1 缺陷综合征(GLUT 1-DS)是SLC2A1基因变异所致,发病机制为血液中的葡萄糖不能转运至大脑功能,而KDT产生的酮体可以代替葡萄糖产能,是该病治疗的首选[6]。一项多中心临床研究指出,KDT治疗18例GLUT1-DS患儿安全有效,仅1例SLC2A1基因错义变异(c.377G>A)的患儿生酮治疗无效[1]。本研究2例GLUT1-DS患儿生酮饮食疗效显著,扩展了GLUT1-DS生酮治疗有效的基因型。
Dravet 综合征患儿被建议可早期进行生酮饮食治疗,文献报道KDT 治疗Dravet 综合征患儿的总体有效率中位数为65%[7]。本研究中纳入了5 例SCN 1 A基因变异所致的Dravet 综合征的患儿,有效率为60%,与文献报道相似。其中c.3733 C>T、c.580 G>A 两个错义变异在多篇文献中报道,为热点变异,但其KDT的疗效尚未报道,本研究中这2例患儿KDT 均效果欠佳,将为更多携带这2 种热点变异的患儿提供治疗方面的指导。例4患儿KDT前发作频率为1周2次,KDT后发作频率降为半个月至1个月1 次,但因仍有发作,且为全身强直阵挛发作,后又加用吡仑帕奈治疗,目前患儿癫痫无发作已达1年。文献报道,吡仑帕奈治疗Dravet综合征的有效率为62.5%~66.7%,也有部分患儿达到癫痫无发作[8]。故对于KDT治疗效果欠佳的患儿可尝试吡仑帕奈治疗,本研究另1例患儿(例8)也已经开始引入吡仑帕奈,疗效有待随访。本研究及既往报道[9-10]的SCN1A基因变异的患儿变异形式各异,包括无义变异、错义变异及移码变异,但KDT 的疗效与此并不相关,同文献[2]报道一致。
SCN2A基因变异导致的癫痫表型谱较广,可导致多种发育性癫痫性脑病(DEE)。文献报道,3个月内发病的DEE患儿,SCN2A的错义变异通常引起功能增强,应用钠离子通道阻滞剂有效;3个月后发病的DEE往往是功能缺失,对钠离子通道阻滞剂效果欠佳[11]。本研究中,结合例9患儿的起病年龄、发作形式和文献报道,推测其SCN2A基因的错义变异为功能增强可能性大,故在初始治疗阶段,曾给予奥卡西平治疗,但患儿癫痫发作反而有增多趋势,后撤下奥卡西平后,予丙戊酸钠+托吡酯足量治疗,患儿发作仍无改善。最终在其2 月龄时启动KDT 治疗,在1周内迅速达到无癫痫发作,随访半年余,患儿癫痫控制可,发育也有改善。通过复习既往文献报道的SCN2A基因变异的患儿,3个月内发病的KDT治疗有效的比例高于3 个月后发病的[11-16]。但值得注意的是也有文献报道3 个月内起病的患儿KDT 治疗无效,后予钠通道阻滞剂苯妥英应用后达到癫痫无发作[11]。但苯妥英治疗窗窄,不良反应大,治疗过程中需密切监测血药浓度。文献报道KDT 可使多不饱和脂肪酸合成增加,可调控电压门控钠通道[16]。既往文献报道中的大部分SCN2A基因变异的患儿,KDT疗效与钠离子通道阻滞剂(SCBs)一致,推测KDT代谢产物是否可以阻滞钠离子通道,但也有几点不符:①部分患儿KDT疗效与SCBs不一致;②在数例SCN2A基因变异功能增强的患儿中,KDT治疗无效;③SCN 1 A基因变异所致Dravet 综合征患儿多为功能缺失,但KDT治疗也有效。故KDT治疗钠离子通道基因变异相关患儿的机制还有待进一步发掘。
磷脂酰肌醇聚糖A 类(PIGA)基因编码蛋白磷脂酰肌醇N-乙酰氨基葡萄糖基转移酶亚基A,是糖基磷脂酰肌醇-甘氨酸(PIG)酶亚类的一个成员。其变异可引起多发先天畸形-肌张力低下-癫痫综合征(MCAHS)2型。MCAHS2表型的严重程度与残余PIGA酶活性的量相关。本研究中例10患儿相对表型较轻,但存在显著的发育迟缓,且癫痫发作药物难以控制。北大医院曾报道了1例与本报道有相同PIGA基因变异的患儿,该患儿表现为面容畸形、先天性巨结肠,同样有局灶性发作和癫痫持续状态,给予维生素B6(100 mg/d)治疗后癫痫发作改善不明显[17]。本研究中例10患儿KDT后9月内仅有一次局灶性发作,持续数秒即缓解,且曾因抽搐频繁发育倒退不能行走,KDT 后能够再次独立行走。既往文献曾报道了1例PIGA基因变异(c.535A>T)的患儿,予苯巴比妥、左乙拉西坦、托吡酯治疗后无效,KDT后达到癫痫无发作[18]。有关KDT治疗的机制,文献中提到遗传性糖基化缺陷癫痫的发病机制之一是由于细胞内γ-氨基丁酸(GABA)的缺失[19],而KDT的作用方式之一是促进GABA的合成和释放。
本研究中例1患儿KIF1A的基因变异(c.296C>T)也是1个热点变异,既往文献已报道7例,表型包括小头畸形、痉挛性截瘫、显著的全面性发育迟缓、视神经病变、难治性癫痫、脑萎缩等,这些临床特征构成了PHEO综合征或PHEO样综合征[20]。本例患儿使用包括KDT在内的多种抗癫痫治疗方案,疗效仍然很差,该病预后很差,目前缺乏有效的治疗手段,但对疾病及致病基因的认识可以帮助产前咨询及将来寻求疾病治疗的突破口。
本研究中有70%的患儿出现不良反应,但均能耐受,未影响KDT的治疗。但既往有报道12%的患者因不良反应而需停止KDT[21]。故筛选合适的患者进行治疗仍具有一定的临床价值。本研究发现KDT对携带有SLC2A1、SCN2A、PIGA、SCN1A部分位点变异的患儿有不错的疗效,这些结果将为考虑早期、有针对性的KDT治疗提供合理的依据。二代测序技术的发展可协助早期明确遗传性病因,而不断发现新的致病基因,并总结其与KDT 疗效的关系,对实现个体化的精准治疗有重要意义。
