固相萃取一步式净化-超高效液相色谱-串联质谱法同时测定畜禽肉中46种兽药的残留量
2022-12-27周雯鹂马腾洲宓捷波程云辉
周雯鹂,马腾洲,清 江,宓捷波,王 飞,许 宙,程云辉,丁 利*
(1.长沙理工大学 食品与生物工程学院,长沙 410114; 2.上海海关 工业品与原材料检测技术中心,上海 200135;3.天津海关 动植物与食品检测中心,天津 300461)
随着我国人民生活水平的提高,人们对于食品安全要求也越来越高,农兽药残留等食品安全问题受到人们的重点关注。在养殖业的现代化和规模化过程中,兽药为降低畜禽发病率与死亡率起到了十分显著的作用,但是一些违禁药物,由于用药疗程较长、剂量过大等,在畜禽机体中没有分解代谢完全,沉积在机体内形成残留[1]。滥用、误用以及不遵守休药期等现象常有发生,导致畜禽产品的食品安全问题日趋严重,严重影响人类的健康[2]。由于畜禽产品中兽药残留超标,我国许多农产品被拒绝进口的事件时有发生,造成我国农产品经济的巨大损失。喹诺酮类、磺胺类、大环内酯类和苯并咪唑类兽药是畜禽养殖行业中常用的抗菌、抗球虫及抗原虫药物[3-4]。滥用这些药物会造成禽兽肉及其制品的污染,且由于食物链的累积,最终将对人类造成危害,如会使机体产生耐药性及过敏反应等,严重的将致突变、致畸形、致癌[5]。因此,喹诺酮类、磺胺类、大环内酯类和苯并咪唑类兽药是我国国内以及进出口肉制品品质监控中的必检项目。目前,国际食品法典委员会(CAC)、欧盟、美国、日本以及我国针对不同种类动物源性食品中的十几类数百种兽药都制定了残留限量标准,大多低至μg·kg-1级水平[6]。因此,建立一种简便易行、快速灵敏、可同时分析畜禽产品中常见的多种兽药的方法非常必要,对保障食品安全具有重要意义。
兽药的主要测定方法有酶联免疫法[7]、高效液相色谱法[8]及超高效液相色谱-串联质谱法(UHPLC-MS/MS)[9-10]。常见的前处理方法有分散固相萃取法[11]、加速溶剂萃取[12]、固相萃取法[13]以及超临界流体萃取法[14]等。UHPLC-MS/MS可以同时检测多种物质,无需复杂的色谱分离过程,分析快速,灵敏度与准确度更高。固相萃取法较其他方法而言,净化效果好,操作更为简单,商品化的固相萃取小柱更适用于快速分析。同时测定多类兽药残留的方法已有报道,如文献[15]采用改进的Qu ECh ERS提取净化鸡肉、鸡肝、猪肉、猪肝样品中的四环素类、磺胺类、喹诺酮类和金刚烷胺类共4类29种禁限用兽药,并通过UHPLC-MS/MS检测;文献[16]采用陶瓷均质子搅拌提取,Qu ECh ERS净化,并结合液相色谱-串联质谱法测定奶酪中大环内酯类、磺胺类、喹诺酮类和四环素类共50种兽药残留量。但是,在对多种兽药残留的检测研究中,苯并咪唑类兽药的检测报道较少,且未见有磺胺类、喹诺酮类、大环内酯类和苯并咪唑类这4类必检兽药残留的同时检测分析方法。对于前处理方法而言,传统的Qu ECh ERS净化小柱通常为粉状填料,提取过程需要在加入溶液后振荡离心,研究表明该方法比较适合含水量大的蔬菜、水果中农药的提取,对动物源性食品中兽药提取效果较差[17],通常需要对Qu ECh ERS方法进行改进。
本工作采用SHI MSEN Qvet-NM固相萃取柱对畜禽肉中喹诺酮类、磺胺类、大环内酯类和苯并咪唑类共46种必检兽药进行了一步式净化前处理,提出了简单、快速的UHPLC-MS/MS的高通量检测方法。该方法无需传统固相萃取法的淋洗、洗脱等步骤,成功应用于实际猪肉、鸡肉样品中兽药残留量的定量分析,为畜禽肉中兽药残留监管提供了技术支撑。
1 试验部分
1.1 仪器与试剂
AB Sciex 6500型超高效液相色谱-串联质谱仪;CF16RN型离心机;AL204-IC型分析天平;HS501型振荡器;ELGA型超纯水仪;SHI MSEN Qvet-NM固相萃取柱(500 mg/6 mL)。
单标准储备溶液:1.0 g·L-1,准确称取一定量的各兽药标准品,分别用甲醇溶解并定容,配制成质量浓度为1.0 g·L-1的单标准储备溶液,置于-18℃下避光保存。
混合标准溶液:移取适量的单标准储备溶液,混合,用甲醇稀释并配制成磺胺类质量浓度为1.6 mg·L-1,喹诺酮类质量浓度为2.0 mg·L-1,大环内酯类及苯并咪唑类质量浓度均为8.0 mg·L-1的混合标准溶液,置于4℃下避光保存。
内标储备溶液:1.0 g·L-1,分别称取适量的内标磺胺喹口恶啉-13C6、环丙沙星-d8、罗红霉素以及氟苯达唑-d3等4种标准品,用甲醇溶解并定容,配制成质量浓度为1.0 g·L-1的内标储备溶液,置于-18℃下避光保存。
混合内标溶液:1.0 mg·L-1,移取各内标储备溶液,用甲醇稀释,配制成质量浓度为1.0 mg·L-1的混合内标溶液,置于4℃下避光保存。
46种兽药包括恩诺沙星、环丙沙星、氧氟沙星、诺氟沙星、培氟沙星、丹诺沙星、氟甲喹、口恶喹酸、萘啶酸、沙拉沙星、二氟沙星、麻保沙星、磺胺氯哒嗪、磺胺二甲氧嘧啶、磺胺甲基嘧啶、磺胺嘧啶、磺胺甲基异口恶唑、磺胺噻唑、磺胺喹口恶啉、甲氧苄啶、磺胺间甲氧嘧啶、4-磺胺-6-甲氧-嘧啶、磺胺二甲异口恶唑、磺胺邻二甲氧嘧啶、磺胺甲噻二唑、磺胺醋酰、磺胺吡啶、磺胺苯吡唑、磺胺胍、阿苯达唑、阿苯达唑砜、2-氨基阿苯达唑砜、噻苯达唑、噻苯达唑酯、奥芬达唑、奥芬达唑砜、5-羟基噻苯达唑、氨基甲苯达唑、阿苯达唑亚砜、氟苯达唑、氧苯达唑、甲苯达唑、羟基甲苯达唑、交沙霉素、泰乐菌素、林可霉素的标准品纯度均大于97%;内标磺胺喹口恶啉-13C6、环丙沙星-d8、罗红霉素以及氟苯达唑-d3的标准品纯度均大于97%;乙腈、甲醇和正己烷均为色谱纯;甲酸为分析纯;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
ZORBAX Eclipse Pl us C18色谱柱(150 mm×4.6 mm,5μm);柱温40℃;进样量5μL;流量0.4 mL·min-1;流动相A为0.1%(体积分数,下同)甲酸溶液;B为乙腈;梯度洗脱程序:0~1.0 min时,A为90%;1.0~7.0 min时,A由90%降至5%,保持1.0 min;8.0~8.1 min时,A由5%跳 转 至90%,保持4.9 min。
1.2.2 质谱条件
电喷雾离子(ESI)源,正离子(ESI+)模式;离子源温度550℃;辅助气1压力344 k Pa;辅助气2压力310 k Pa;多反应监测(MRM)扫描模式。其他质谱参数见表1,其中,“*”代表定量离子。
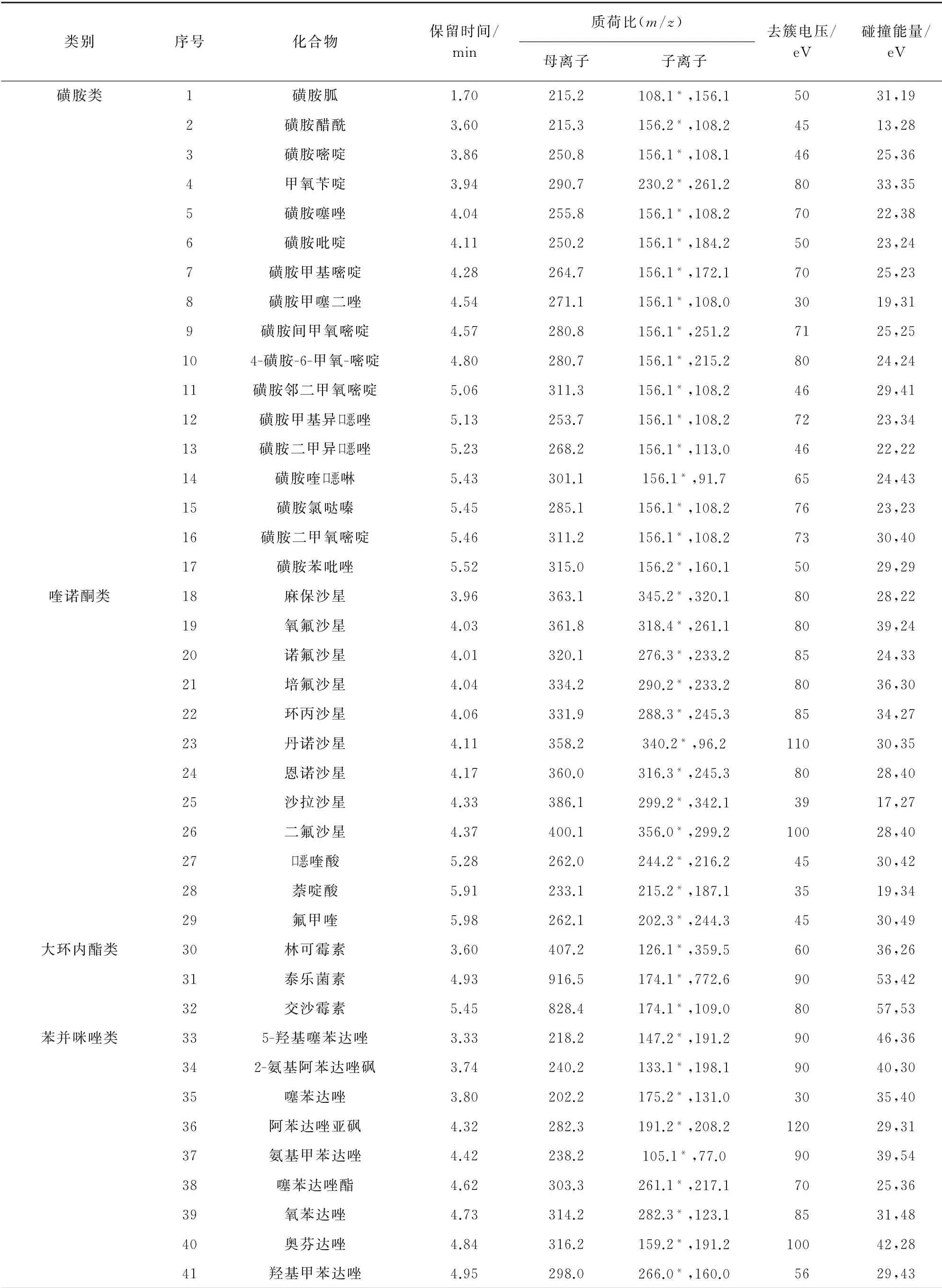
表1 质谱参数Tab.1 MS parameters

表1 (续)
1.3 试验方法
称取5 g猪肉或鸡肉样品,置于50 mL具塞离心管中,加入1 mg·L-1混合内标溶液20μL,并加入20 mL乙腈,振荡混匀,以转速5 000 r·min-1离心5 min,上清液转移至另一个50 mL具塞离心管中。在剩余物中再加入15 mL乙腈,重复提取一次,合并上清液。向提取液中加入15 mL正己烷,混匀,以转速4 000 r·min-1离心2 min,弃去正己烷层,然后再用15 mL正己烷重复上述操作,进行二次脱脂。将提取液过SHI MSEN Qvet-NM固相萃取柱,收集全部流出液,于40℃水浴减压浓缩至近干,用1 mL 30%(体积分数)甲醇溶液定容,过0.22μm有机相滤膜,按照仪器工作条件测定。
2 结果与讨论
2.1 样品前处理条件的优化
2.1.1 提取溶剂
畜禽肉等样品基质中通常含有蛋白质、脂肪以及色素等物质,易干扰测定结果,因此在前处理过程中需要进行脱脂和脱蛋白。乙腈具有良好的沉淀蛋白的能力,可以避免从样品基质中提取过多的脂肪,且4类兽药均为极性较弱的化合物,采用乙腈或者酸化的乙腈提取可以获得较好的提取效果[18]。正己烷是常见的用于脱脂的溶剂,可以有效地去除样品中的脂肪。因此,样品在采用SHI MSEN Qvet-NM固相萃取柱一步净化富集前,先由乙腈振荡离心提取,并加入正己烷脱脂。
2.1.2 净化方法
为了缩短前处理时间,试验比较了Qu ECh ERS分散固相萃取与SHI MSEN Qvet-NM固相萃取柱净化等两种不同前处理方法的净化效果。本方法采用内标法进行回收率校正,但为了更直观地比较两种前处理方法的优劣,在优化时未添加内标,采用兽药的绝对回收率进行比较。猪肉、鸡肉两种样品基质中4类兽药的平均回收率如图1所示。

图1 净化方法对4类兽药平均回收率的影响Fig.1 Effect of purificationmet hods on average recovery of 4 veterinary drugs
由图1可知,采用SHI MSEN Qvet-NM固相萃取柱进行前处理得到的平均回收率明显优于Qu ECh ERS分散固相萃取方法。SHI MSEN Qvet-NM固相萃取柱通过正压或抽负压的方式,使待净化的样品提取液直接通过净化床层,达到杂质去除的目的,与传统的Qu ECh ERS分散固相萃取方法进行比较,可以有效缩短前处理时间,减少溶剂使用。因此,试验选择SHI MSEN Qvet-NM固相萃取柱用于一步式净化。
2.2 色谱条件的优化
为了提高所测化合物的质谱响应,比较了乙腈-0.1%甲酸溶液、甲醇-0.1%甲酸溶液两种流动相体系以及ZORBAX Eclipse Plus C18、ZORBAX SB C18、Sunfire C18等3种色谱柱对4类兽药分离效果的影响。结果显示:甲醇对磺胺类同分异构体的分离较好,但对大环内酯类和苯并咪唑类的分离效果不如乙腈;在3种色谱柱中,4类兽药在ZORBAX Eclipse Pl us C18色谱柱上峰形更加尖锐,分离度最佳。因此,试验最终采用乙腈-0.1%甲酸溶液作为流动相体系,ZORBAX Eclipse Pl us C18色谱柱作为分离柱。
2.3 质谱条件的优化
为优化质谱条件,采用流动注射分析法(FIA)。在调谐模式下,采用一级质谱扫描,调节去簇电压使每个化合物的母离子丰度达到最佳,再由二级质谱扫描各个化合物的特征碎片离子,通过调节碰撞能量优化所有被测分析物碎片离子强度。具体的质谱参数见表1。
2.4 基质效应
采用基质匹配和纯溶剂下相同浓度水平化合物的质谱响应值的比值计算基质效应因子(MF)值,进而评价46种兽药在样品中的基质效应。当纯溶剂与基质匹配下的质谱响应比值大于1时,表示存在基质增强效应;反之则存在基质抑制效应。以猪肉样品为研究对象,具体的基质效应结果见表2。

表2 猪肉基质中46种兽药的MF值Tab.2 MF values of 46 veterinary drugs in pork matrix
结果显示,猪肉基质中46种兽药的MF值为0.65~0.95,均存在基质抑制效应,因此在试验中采用基质匹配的混合标准溶液系列进行定量。
2.5 标准曲线和检出限
取空白样品,按照优化后的提取方法对样品进行处理,根据目标化合物在质谱中响应的强弱,在提取液中加入混合标准溶液与混合内标溶液,配制成基质匹配的混合标准溶液系列,其中,磺胺类质量浓度为0.5,1.0,2.0,10,16μg·L-1,喹诺酮类质量浓度为0.5,1.0,2.0,10,20μg·L-1,大环内酯类和苯并咪唑类质量浓度均为2.5,5.0,10,50,80μg·L-1,4种内标质量浓度均为20μg·L-1。按照仪器工作条件测定基质匹配的混合标准溶液系列,以目标物与内标的峰面积比值为纵坐标,目标物的质量浓度为横坐标进行线性回归。所得线性参数见表3。
以3倍信噪比(S/N)计算检出限(3S/N),并与欧盟规定的肉制品中兽药残留限量进行比较,结果见表3。

表3 线性参数与检出限Tab.3 Linearity parameters and detection limits

表3 (续)
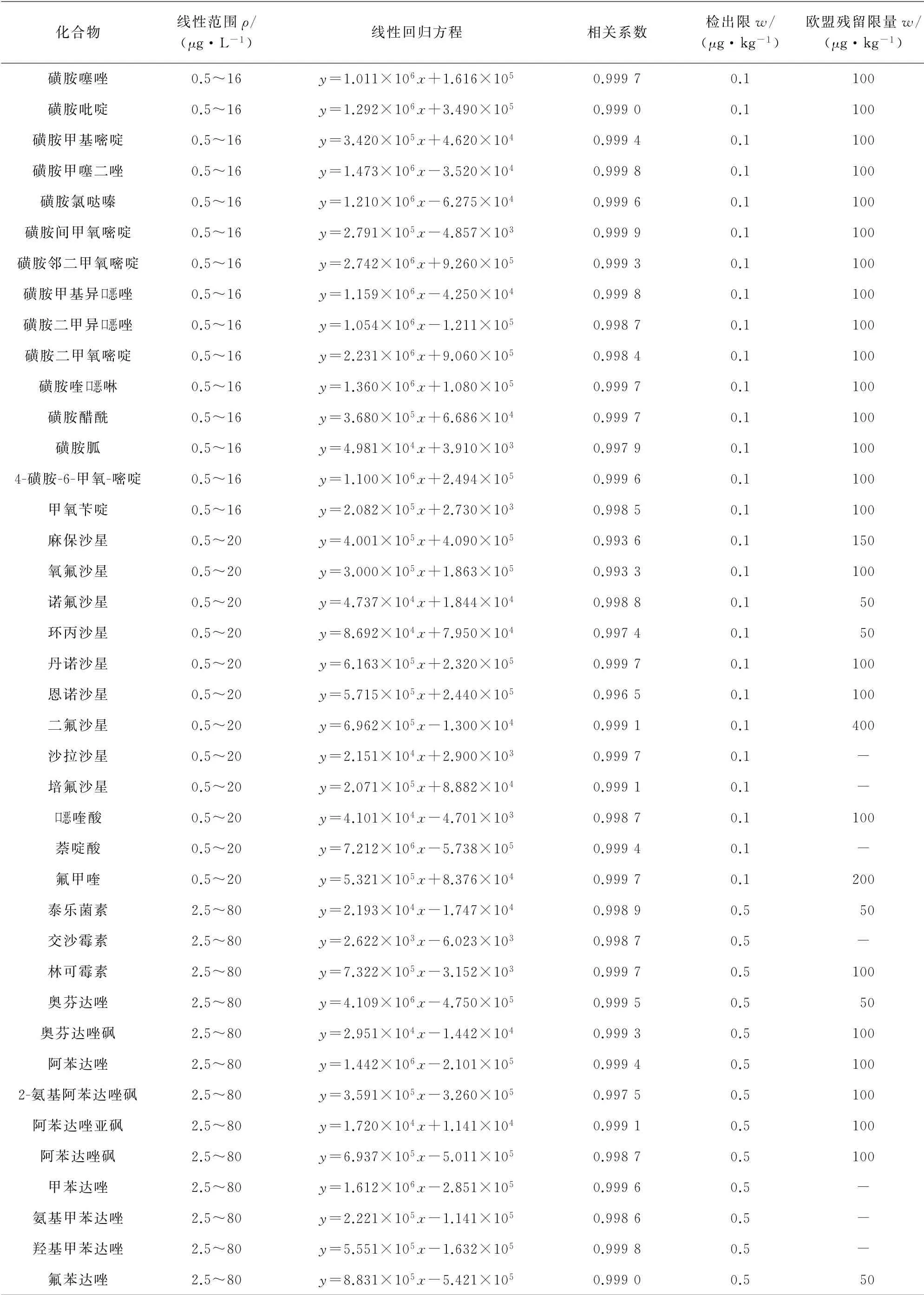
表3 (续)
由表3可知,兽药的检出限远低于欧盟规定的肉制品中兽药残留限量(禁用的兽药除外)。
2.6 精密度和回收试验
对空白猪肉基质进行加标回收试验,其中喹诺酮类、磺胺类兽药的加标量分别为0.2,0.4,2.0μg·kg-1(相当于1.0,2.0,10μg·L-1),苯并咪唑类、大环内酯类兽药的加标量分别为1.0,2.0,10μg·kg-1(相当于4.0,10,50μg·L-1),每个加标水平制备6个平行样,计算回收率和测定值的相对标准偏差(RSD),结果见表4。

表4 精密度和回收试验结果(n=6)Tab.4 Results of tests for precision and recovery(n=6)
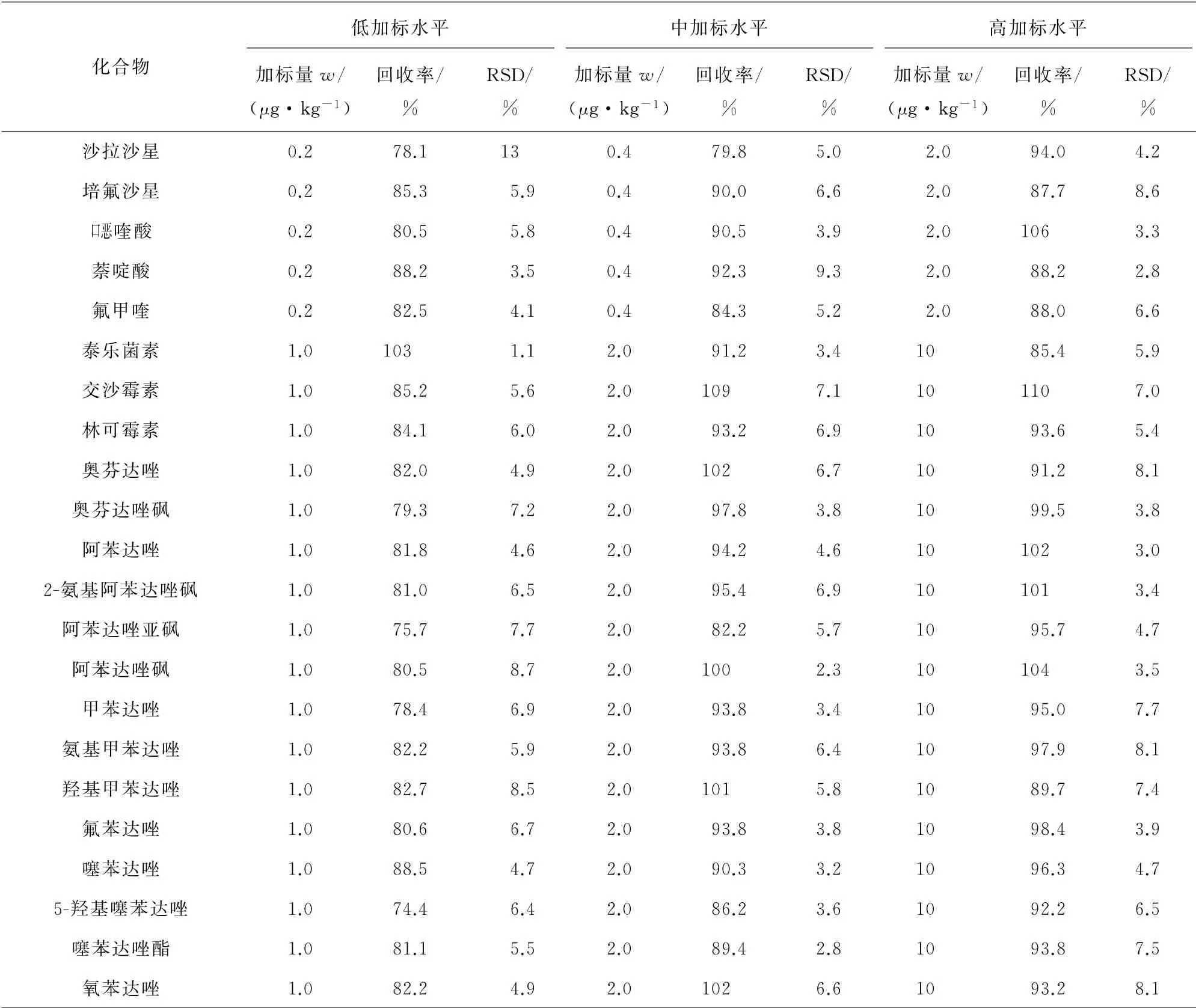
表4 (续)
结果表明,46种兽药的回收率为72.9%~110%,测定值的RSD均小于15%。说明该方法准确度高、精密度好,符合兽药分析要求。
2.7 样品分析
采用所建立的方法和国家标准GB/T 40462-2021《有机肥料中19种兽药残留量的测定 液相色谱串联质谱法》分别对8批市售猪肉、鸡肉进行测定。结果显示,8批样品中上述化合物均未检出,本方法测定结果与国家标准方法一致,符合兽药残留测定的标准。
本工作提出了固相萃取一步式净化-UHPLCMS/MS同时测定畜禽肉中磺胺类、喹诺酮类、大环内酯类和苯并咪唑类共4类46种兽药残留量的方法。该方法采用乙腈两次振荡提取、正己烷重复脱脂、SHI MSEN Qvet-NM固相萃取柱一步式净化后,再经UHPLC-MS/MS进行定量分析。与现行国家标准及行业标准方法相比,该方法更简便、快速,能实现同时分析多类兽药残留,可为畜禽肉中兽药多残留的定性及定量分析提供技术依据。
