T7噬菌体尾丝蛋白随机进化文库的构建
2022-12-24洪伟鸣李睿婷郭子杰左伟勇
洪伟鸣,李睿婷,郭子杰,徐 海,左伟勇,张 亮,宋 亮
(江苏农牧科技职业学院 江苏省兽用生物制药高技术研究重点实验室,江苏 泰州 225300)
噬菌体是一类寄生在原核生物,如细菌、真菌、放线菌或螺旋体等宿主细胞内的病毒,在发现之初就被用于细菌感染性疾病的治疗[1]。随着人类进入抗生素时代,噬菌体治疗逐渐淡出公众的视野。近年来由于抗生素的滥用,导致耐药菌的不断出现,细菌的耐药性已然成为全球范围内的公共卫生问题,噬菌体作为细菌的天然杀手再次受到关注[2-3]。
噬菌体对其宿主的识别具有严格的特异性,不同种、甚至不同分离株常常表现出差异较大的噬菌体敏感性[4]。宿主谱较窄已然成为限制噬菌体抗菌发展与运用的主要障碍之一。鸡尾酒疗法和广谱噬菌体分离被认为是克服这一障碍最为经济有效的途径。将不同宿主识别谱的噬菌体配伍成混合制剂即为鸡尾酒疗法,格鲁吉亚的Eliava噬菌体治疗中心已成功运用该方法数十年[5-6]。但该方法也存在一定的被动性,Eliava中心需定期更新鸡尾酒制剂的噬菌体构成,以保证治疗针对性与有效性。随着分子生物学技术、基因编辑以及各种生物组学的快速发展,人们对噬菌体的认知已经超出形态学观察及生理生化活性鉴定的范畴,逐步从被动筛选自然突变株噬菌体,向主动改造或创造新噬菌体转变。其中,CRISPR-Cas[6]、Bacteriophage recombinant of electroporated DNA(BRED)[7]等技术正在被广泛地用于噬菌体改造。Scholl等[8]通过基因改造使T7噬菌体能够表达一种唾液酸酶,利用该酶消化大肠杆菌产生的K1荚膜,进而拓展了T7噬菌体宿主谱。乐率[9]将PaP1噬菌体的尾丝蛋白替换成JG004噬菌体对应蛋白,使得新构建的PaP1噬菌体获得感染JG004宿主的能力,但丧失对原有宿主的裂解性。T4噬菌体感染大肠埃希菌以及亲缘关系近的志贺氏菌,Tétart等[10]将噬菌体SV76.3和Mi基因组成的片段置换T4噬菌体尾丝基因的同源结构,构建的杂合噬菌体不仅能识别天然宿主,也能够识别假结核耶尔森菌。
T7噬菌体是侵染大肠埃希菌的小型烈性噬菌体,其病毒颗粒有二十面体头部和非常小的尾,二者之间由头尾颈圈连接。T7噬菌体gp17基因编码533氨基酸的尾丝蛋白(p17),每个噬菌体含有6条尾丝蛋白(图1-A),为同源三聚体结构,主要负责识别宿主、启动感染。决定T7噬菌体宿主识别的区域位于最末端的球状区,结构学解析表明,该区域有4个暴露的柔性loop环(BC-、DE-、FG-和HI-loop)(图1-B),该区域氨基酸组成的改变导致宿主识别谱的变化,是主要的宿主识别区域[11]。易错PCR(error-prone PCR,EP-PCR)通过改变常规PCR的扩增条件来调整反应过程中的错配频率,进而获得随机突变的DNA群体,其操作简便,特别适用于构建较小基因片段的突变文库。本研究利用EP-PCR技术对T7噬菌体尾丝蛋白羧基末端基因序列进行随机突变,拯救T7噬菌体尾丝蛋白随机进化文库,为后续筛选相对广谱噬菌体或者特定病原菌噬菌体奠定基础。
1 材料与方法
1.1 试验材料
噬菌体与质粒:T7 Select 415-1b噬菌体(GenBank accession number V01146噬菌体为骨架改造)拯救试剂盒购于Merck公司,pMD19-T simple载体购于宝生物公司。
主要试剂:限制性内切酶、TaqDNA聚合酶、T4 DNA连接酶、Diversify PCR Random Mutagenesis Kit均为宝生物公司产品;胶回收试剂盒为QIAGEN公司产品;其余试剂均为分析纯。
主要仪器:台式离心机购自美国Beck-man公司;BTX电转化仪、PCR仪购自宝生物公司;Geldoc-It Imaging System购自美国UVP公司。
1.2 T7噬菌体基因组提取与酶切
采用苯酚-氯仿抽提的方法提取T7噬菌体基因组[12],100 mL T7噬菌体培养液8 000 r·min-1离心15 min,去除裂解细菌的碎片,收集上清液。加入终浓度为1 μg·mL-1的RNase A、DNaseⅠ,37 ℃反应1 h,以1∶4体积比加入PEG-NaCl溶液(20%PEG-8000,2.5 mol·L-1NaCl),摇匀溶解后,放置4 ℃过夜。次日,12 000 r·min-1离心30 min并收集沉淀。用5 mL SM缓冲溶液重悬沉淀,加入50 μL 10%SDS、50 μL 0.5 mol·L-1EDTA,于60 ℃消化30 min。加入等体积的DNA提取液(苯酚/氯仿/异戊醇体积比 25∶24∶1),抽提2次,转移上清液,加入等体积的乙醇混匀,-20 ℃放置1 h,12 000 r·min-1离心10 min,收集沉淀并用1 mL预冷的70%乙醇洗涤沉淀2次,12 000 r·min-1离心10 min,弃上清,沉淀于室温下自然晾干,然后溶于100 μL TE溶液,即为提取的T7噬菌体DNA。取10 μL基因组经SfiⅠ单酶切,0.5%琼脂糖凝胶电泳鉴定后切胶回收约33 kb片段。
1.3 常规PCR扩增
以T7噬菌体基因组为模板:用引物F1/R1扩增尾丝蛋白羧基末端(Tail fiber carboxy-terminal,TF-ct)33 308~33 663位约350 bp基因序列。反应程序为:94 ℃ 30 s;55 ℃ 30 s;72 ℃ 30 s,30个循环。在F1引物5′端引入SfiⅠ酶切位点,R1引物5′端引入SphⅠ酶切位点。用引物F2/R2扩增基因组33 663~37 314位约4 000 bp基因序列。反应程序为:94 ℃ 30 s;52 ℃ 45 s;72 ℃ 2 min,30个循坏,并在F2引物5′端引入SphⅠ酶切位点。PCR产物经1.0%琼脂糖凝胶电泳鉴定,SphⅠ单酶切4 000 bp片段,切胶回收,-20 ℃保存备用。引物序列见表1。
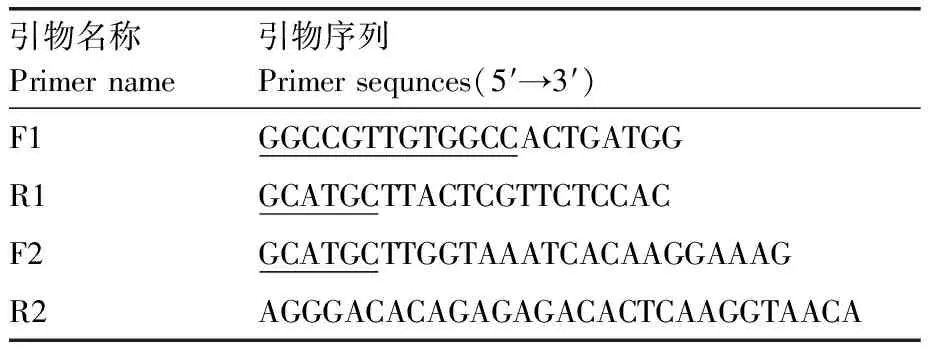
表1 文库构建及鉴定引物
1.4 易错PCR扩增
以回收的TF-ct基因片段为模板,用F1/R1引物进行EP-PCR扩增。反应体系为50 μL,其中包含模板(1 ng·μL-1,1 μL)、引物(10 μmol·L-1,1 μL)和TITANIUMTaq酶(1 μL);比较3种缓冲条件(buffer condition,BC)下扩增效率和突变率的差异:BC9(buffer,5 μL、8 mmol·L-1Mn2+,4 μL、2 mmol·L-1dGTP,5 μL)、BC6(buffer,5 μL、8 mmol·L-1Mn2+,4 μL、2 mmol·L-1dGTP,2 μL)、BC3(buffer,5 μL、8 mmol·L-1Mn2+,2 μL、2 mmol·L-1dGTP,1 μL)。EP-PCR反应条件:94 ℃预变性30 s;94 ℃变性 30 s,68 ℃退火延伸1 min,25个循环。1%琼脂糖凝胶电泳鉴定EP-PCR扩增产物,并切胶回收。
1.5 质粒文库的构建
将突变的TF-ct基因以3∶1的物质的量之比与pMD19-T simple连接,转化大肠埃希菌DH5α感受态细胞。转化产物涂布在含有Amp+/IPTG/X-gal LB固体培养基的培养皿上,37 ℃过夜。统计菌落总数和蓝色菌落数,计算质粒文库的容量和重组率[13]。质粒库容量=菌落总数×转化产物量;重组率(%)=(菌落总数-蓝色菌落数)/菌落总数×100。剔除蓝色菌落,收集培养皿中所有菌落,LB液体培养基重悬菌落,提取质粒经SfiⅠ、SphⅠ双酶切鉴定,回收350 bp目的片段。
1.6 噬菌体文库的构建
将SfiⅠ单酶切回收的33 kb基因组片段、SfiⅠ和SphⅠ双酶切回收的随机突变TF-ct片段以及SphⅠ单酶切回收的4 000 bp片段以1∶1∶1的物质的量之比进行连接(图1-C)。取5 μL连接产物与25 μL包装蛋白混合,25 ℃反应2 h,反应体系中加入270 μL LB液体培养基终止反应,即为拯救的噬菌体文库[13]。取5 μL包装产物10倍梯度稀释并与200 μL过夜培养的T7噬菌体宿主细菌BL21混合,经双层琼脂夹心法测定噬菌体滴度;挑取单噬斑,用引物F1/R1进行PCR检测目的基因插入情况。每组随机挑选5个噬斑PCR阳性产物进行序列测定,采用DNAStar序列分析软件对测序结果进行比对分析。分别统计目的基因中突变的碱基数及对应的氨基酸突变数,按照突变率(%)=突变碱基数(个)/目的基因大小(bp)×100计算突变率。将剩余的295 μL包装产物转接100 mL对数生长期BL21,37 ℃摇床培养3 h至宿主细菌完全裂解,PEG-NaCl沉淀法回收噬菌体,即为构建的T7噬菌体尾丝蛋白随机进化文库。
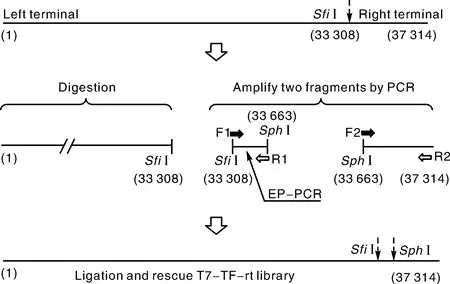
图1 定向进化文库构建原理及流程
1.7 单克隆噬菌体吸附力比较
从已测序的15个单克隆噬菌体中随机挑选EP3、EP6、EP10、EP12和EP13这5个在loop区域有点突变的克隆进行纯培养,用于比较宿主吸附能力的差异。将宿主细菌BL21培养至对数生长期,并在预热的LB培养基稀释至1×107CFU·mL-1。以5 mL每管分装BL21细菌,往其中分别接种单克隆噬菌体至浓度为1×105PFU·mL-1,并于37 ℃继续孵育5 min。取200 μL孵育混合物立即离心分离游离噬菌体和吸附在BL21表面的噬菌体;另取200 μL孵育混合物不做离心处理。分别测定离心后上清中游离噬菌体的滴度(Nf)和未离心处理混合物中的噬菌体滴度(Nt)。计算噬菌体吸附率α=-0.2[ln(Nf/Nt)][14]。
2 结果与分析
2.1 基因片段的制备
从T7噬菌体培养液中成功提取到高纯度T7噬菌体基因,电泳显示基因组条带单一、大小符合预期;经SfiⅠ单酶切可将线性T7噬菌体基因组分成约33 kb左侧片段以及约4 000 bp右侧片段(图2-A)。以提取的基因组为模板,F1/R1引物扩增出350 bp的T7噬菌体尾丝蛋白羧基末端基因片段(图2-B),F2/R2引物扩增出尾丝蛋白基因下游约4 000 bp基因片段(图2-C)。
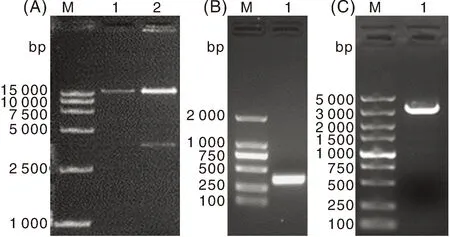
A:T7噬菌体基因组提取及酶切;M,DL15000 DNA marker;1,T7噬菌体基因组;2,SfiⅠ单酶切T7噬菌体基因组。B:PCR扩增T7噬菌体尾丝蛋白羧基末端基因片段;M,DL2000 DNA marker;1,TF-ct基因片段。C:PCR扩增T7噬菌体尾丝蛋白下游末端基因片段;M,DL5000 DNA marker;1,尾丝蛋白下游末端基因片段。
2.2 TF-ct基因片段的随机突变及质粒文库构建
为了有效扩增TF-ct基因片段同时保证获得足够的突变率,根据突变试剂盒使用说明,选择3个缓冲条件进行EP-PCR扩增。随着扩增体系中Mn2+、dGTP含量的逐渐下降,从图3-A可以看出,目的条带的亮度逐渐升高,说明下调扩增体系中Mn2+及dGTP的含量有助于提高扩增效率。分别回收3个缓冲条件下扩增的TF-ct基因片段,与pMD19-T simple载体连接,转化DH5α感受态细胞,构建质粒文库。提取文库质粒,经SfiⅠ和SphⅠ双酶切,获得与预计大小相符的目的条带(图3-B),说明质粒文库构建成功。重复构建质粒文库3次,其库容分别为1.25×107、2.33×107、3.46×107CFU·mL-1,通过统计蓝白斑比率发现3个质粒文库的重组率均高达90%以上。

A:EP-PCR扩增TF-ct 基因片段;M,DL5000 DNA marker;1~3,BC9、BC6和BC3缓冲条件下扩增TF-ct基因片段。B:双酶切鉴定质粒文库;M,DL5000 DNA marker;1~3,BC9、BC6和BC3质粒文库双酶切鉴定。
回收SfiⅠ和SphⅠ双酶切条带,用于T7噬菌体尾丝蛋白定向进化文库的构建。
2.3 随机进化文库的构建及鉴定
按照图1-C中的构建流程,将33 kb的T7噬菌体基因左侧片段、3个质粒文库中回收的TF-ct片段以及尾丝蛋白下游至基因右侧的片段,以等物质的量之比进行连接,通过包装蛋白包装拯救T7噬菌体尾丝蛋白随机进化文库。测定BC9、BC6和BC3噬菌体文库的滴度分别为1.37×107、5.52×107和8.26×107CFU·mL-1,并随机挑选5个噬斑进行PCR鉴定,从图4-A可以看出,TF-ct基因均成功插入到设计位点。对15个PCR产物进行序列测定,通过与原始序列相比对发现(图4-B),随机挑选的15个克隆均有随机突变位点,突变率高达100%。
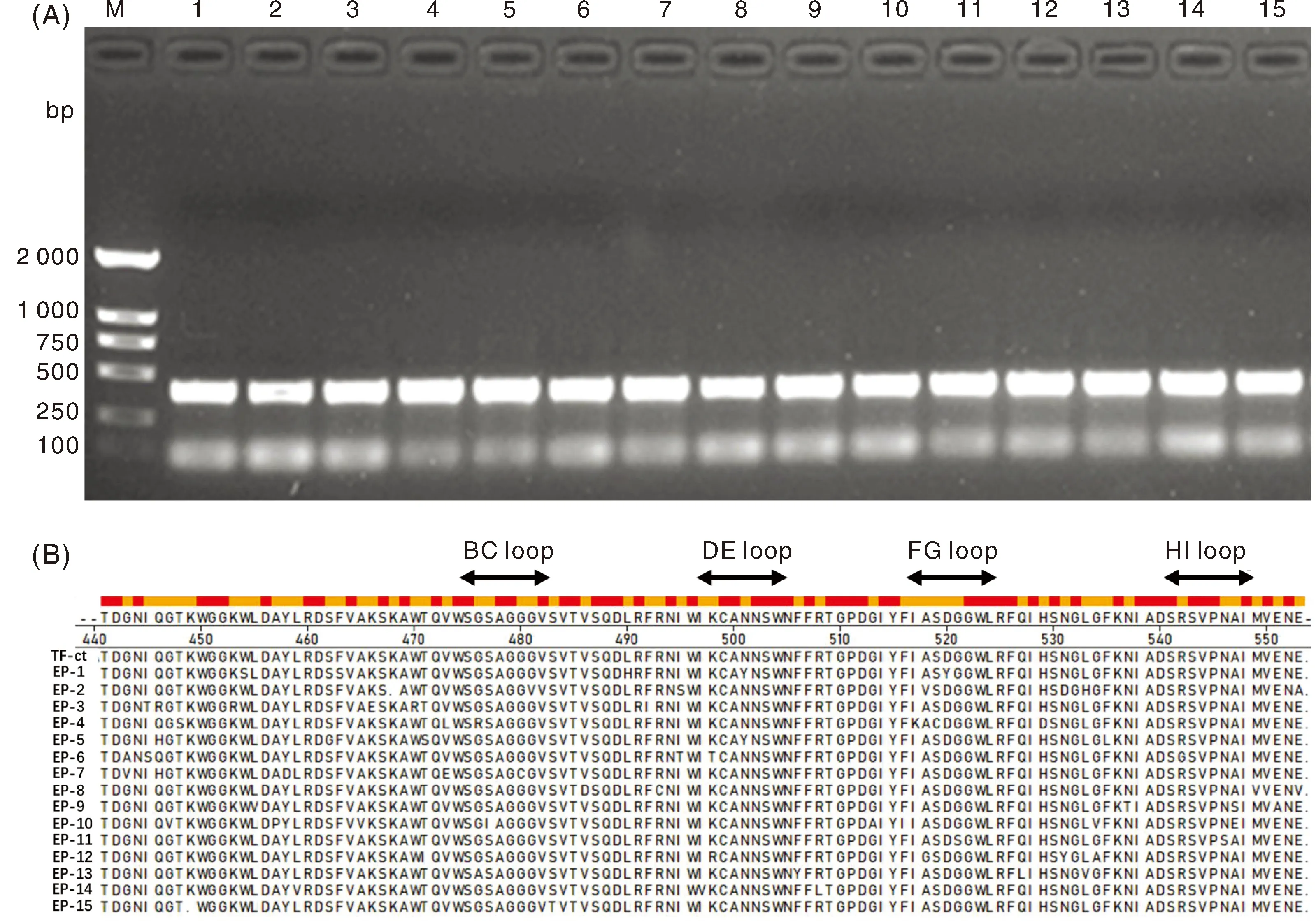
A:单克隆噬菌体PCR鉴定;M,DL2000 DNA marker; 1~5,BC9噬菌体文库;6~10,BC6噬菌体文库;11~15,BC3噬菌体文库T7噬菌体基因组。B:氨基酸序列分析;15个测序样品的氨基酸序列各需相同,且在对应的4个loop区域均有氨基酸点突变。
2.4 不同缓冲条件下突变率的分析
本研究选择BC9、BC6和BC3三个缓冲条件,并依次降低体系中Mn2+和dGTP浓度,从表2可以看出:随着Mn2+和dGTP浓度的降低,其平均碱基突变数及对应氨基酸突变数均同步下降。
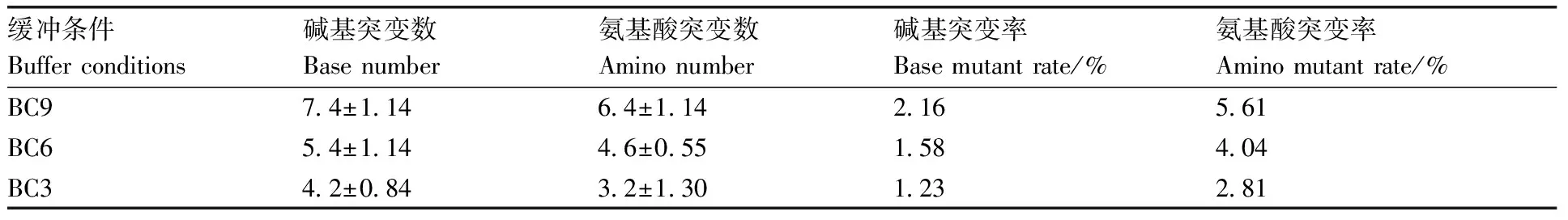
表2 不同BC条件下EP-PCR的突变率统计
2.5 尾丝蛋白突变对吸附能力的影响
分析已测序噬菌体单克隆TF-ct氨基酸序列,挑选EP-4(I 517K)、EP-6(K 498 T)、EP-10(S 477 I)、EP-12(K 498 R)和EP-13(G 476 A)5个在loop区域有点突变的单克隆进行纯培养。通过测定突变株T7噬菌体吸附大肠埃希菌宿主能力,评价尾丝蛋白羧基末端loop区域氨基酸突变对T7噬菌体生物学功能的影响。以T7-wt作为对照,比较吸附能力,从图5可以看出,EP-6、EP-12对应于DE loop的突变降低了噬菌体与宿主的吸附能力,而EP-10、EP-13对应于BC loop的突变,仅EP-13提高了噬菌体与宿主的吸附能力。
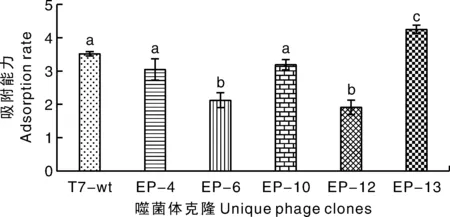
以T7-wt标准株为参照,比较5株尾丝蛋白点突变T7噬菌体吸附宿主大肠埃希菌的效率差异,相同字母标记表示差异不显著,不同字母标记表示差异显著(P<0.05)。
3 结论与讨论
自然进化往往是随机的过程,而实现进化的两个前提是突变与筛选。定向进化则是人为设定标准的选择,很早就被运用于动植物的育种筛选。针对特定蛋白分子的体外定向进化,又被称为实验分子进化,无需事先了解蛋白的空间结构和作用机制,几乎适用于任何蛋白质的分子改造和筛选,这为研究蛋白质的结构和功能提供了一种更为方便、快捷的途径。定向进化的成功实现,需要满足3个方面的条件:合理的进化策略、合适的表达系统以及高通量筛选方法[15]。
在分子生物学飞速发展的今天,定向进化的先驱们建立了许多经典的进化策略和技术,例如Leung团队建立的EP-PCR技术[16]、Willem教授建立的DNA Shuffling[17]技术以及Miguel Alcalde开发的MORPHING[18]技术等,为定向进化研究提供了技术保证。其中,EP-PCR技术是在常规PCR基础上演化而来的,是在扩增过程中引入错误配对,使得扩增产物出现数量众多的点突变,是一种体外诱导DNA序列突变的高效方法。扩增过程中DNA聚合酶保真性、4种dNTP比例、Mg2+离子浓度、Mn2+离子浓度、模板浓度等都是影响错配率的关键因素,优化EP-PCR条件可以确保获得随机度更高的扩增产物。本研究中通过采用3种不同浓度的Mn2+、dGTP进行EP-PCR扩增,获得3个不同突变率的目的片段,从而使尾丝蛋白羧基末端的突变子覆盖最适合的范围,提高了后期文库筛选的得率。
从自然界中筛选相对广谱的噬菌体是一项长期且艰巨的工作,而噬菌体宿主识别谱较窄的特性更是给这项工作增加了难度[19]。T7噬菌体通过尾丝蛋白与宿主细菌表面LPS相作用,每条尾丝独立结合LPS能力很弱且可逆,这样使得T7噬菌体能沿着细菌表面滑动;只有当多条尾丝和LPS相互作用后才能使噬菌体在细菌表面稳定定植,此时基盘正对细菌外膜,这种初步吸附使信号从噬菌体底部传递到头部,从而启动感染的不可逆步骤。决定T7噬菌体宿主识别的区域位于尾丝蛋白最末端的第4个球状区,结构学解析表明,该区域有4个暴露的柔性loop环,是主要的宿主识别区域。Heineman等[20]发现T7噬菌体p17蛋白在FGloop的Asp520突变成Glu和HI-loop的Val544突变成Ala,使其丧失与宿主E.coliB和E.coliK2的结合能力,说明该区域的氨基酸组成对宿主的选择识别至关重要,氨基酸的突变导致其宿主识别的改变。本研究中挑选5个loop区域发生点突变的克隆进行宿主吸附能力的差异比较,发现在DE loop的K498T和K498R突变能够降低T7噬菌体与宿主的吸附能力,而位于BC loop的G476A突变能够提高T7噬菌体与宿主的吸附能力,该发现再次验证了T7噬菌体尾丝蛋白loop区域与宿主识别的相关性。
受到实验室生物材料积累的限制,尚未针对临床细菌样品尤其是畜禽致病性大肠埃希菌开展相对广谱的T7噬菌体筛选。但本研究构建的T7噬菌体尾丝蛋白随机进化文库为后续筛选工作奠定了良好的基础。
