半乳糖凝集素-3在特发性肺纤维化中作用的研究进展
2022-12-19李芳伟万毅新
王 丹,李芳伟,万毅新
特发性肺纤维化(idiopathic pulmonary fibrosis, IPF)是一种慢性、进行性、纤维化性间质性肺疾病,可能由于反复的肺泡上皮细胞损伤和随后的异常愈合所引起,其潜在的发病机制尚未完全明确。IPF的病理表现涉及细胞外基质异常沉积、肌成纤维细胞分化、成纤维细胞增殖而凋亡减少,以及炎症细胞异常浸润等,引起肺实质结构进行性重塑、不可逆转的瘢痕形成,最终导致肺功能下降、甚至衰竭[1]。IPF的治疗目前仍有局限性,药物治疗方面吡非尼酮和尼达尼布对患者有中度疗效,且副作用明显、价格昂贵,肺移植因供体稀缺、排斥反应等受到限制[2]。目前IPF患者的生存期低、病死率高,迫切需要更安全有效的治疗方案。
半乳糖凝集素-3(Galectin-3, Gal-3)与肺、心、肝等多种脏器纤维性疾病有关[3]。在肺纤维化动物模型中以及IPF患者中发现Gal-3可能是IPF发病的重要介质和有效的治疗靶点[4-6],但具体作用机制尚不明确。本文现就Gal-3及其抑制剂在IPF中的作用研究进展进行阐述,以期提供一定的临床价值。
1 Gal-3
凝集素是在植物和动物中发现的,可与糖缀合物特异性结合的碳水化合物结合蛋白。半乳糖凝集素属于动物凝集素家族,是一组水溶性、非糖基化的球状蛋白,包含1个或2个130个氨基酸长的保守碳水化合物识别结构域,因对β-半乳糖苷衍生物的亲和力及共有的氨基酸序列这两种特点,区别于其他动物凝集素[7]。半乳糖凝集素在细胞质中合成,在细胞核、细胞质以及细胞外基质中发挥作用,但因没有蛋白质通过“内质网-高尔基体”经典分泌途径分泌所必需的信号序列,是通过细胞主动分泌方式分泌[7]。目前,哺乳动物中发现有15种半乳糖凝集素,分原型、嵌合型、串联重复型三种类型。Gal-3是唯一的嵌合型,由人类14号染色体上的LGALS3基因编码合成,具有一个羧基末端、中间的胶原样重复序列和一个氨基末端[8]。在成人中,Gal-3普遍分布于造血组织、淋巴结、呼吸道等组织,既存在于细胞内、细胞外,也可以膜分子的形式存在,参与细胞信号调节、增殖、炎症、免疫、血管生成、癌变、自噬调节等多种病理、生理过程[9]。目前研究发现,Gal-3可能主要通过促进炎症、促进组织纤维化等过程参与肺纤维化的发生、发展。
2 Gal-3在IPF中的作用
2.1 Gal-3通过促进炎症反应参与IPF的发生目前认为IPF主要是由肺泡和气道上皮细胞受损引起[10]。组织在受损后会启动一系列包括早期急性炎症的修复过程,以恢复器官的完整性,这时引起白细胞在受损组织中渗透、激活和积聚,浸润的白细胞不仅能激活成纤维细胞,还能自行产生胶原促进组织纤维化[11]。虽然炎症对组织再生及愈合至关重要,但持续的炎症可能会导致组织损伤、纤维化,甚至器官衰竭。Gal-3在髓系细胞(包括单核细胞、巨噬细胞、树突状细胞和中性粒细胞)中高表达,肺泡巨噬细胞、上皮细胞和肺泡细胞是肺内Gal-3的主要来源[6,12]。研究发现,Gal-3在调节先天免疫以及促进急性和慢性炎症中发挥重要作用。Gal-3能激活T淋巴细胞、B淋巴细胞、单核巨噬细胞和中性粒细胞[13-15]。在急性炎症反应中,Gal-3促进单核细胞及巨噬细胞趋化、中性粒细胞清除及肥大细胞脱颗粒,并对中性粒细胞的凋亡有调理作用[16-17]。Gal-3作为模式识别受体和损伤相关模式分子促进炎症小体的组装,炎症小体产生的白细胞介素-1(interleukin-1, IL-1)和IL-18促进炎症反应,并激活未折叠的蛋白反应,未折叠的蛋白反应通过增强核因子-κB(nuclear factor-kappa B, NF-κB)和其他途径来放大炎症反应[18]。Gal-3通过激活细胞外ERK、AKT和JAK-STAT1信号通路,导致流感病毒和肺炎链球菌同时在感染期间促炎细胞因子的释放失调[19],并通过巨噬细胞NLRP3炎症体激活促进宿主炎症反应,从而增强H5N1禽流感病毒的致病作用[20]。Toll样受体4(Toll-like receptors 4, TLR4)可促进涉及干扰素相关基因、白介素、趋化因子以及Gal-3表达的强烈炎症反应,而Gal-3又可激活TLR4,两者相互作用促进肺部炎症进展[21]。Gal-3还可与细菌、病毒和真菌表面的聚糖结合,在识别病原体方面具有潜在作用[22]。最新研究发现,选择性髓系细胞耗竭可降低小鼠肺组织中的(主要是中性粒细胞和肺泡巨噬细胞)Gal-3水平,继而减少中性粒细胞募集和外渗,减轻脂多糖诱导的急性肺损伤以及博莱霉素诱导的肺纤维化[6,23]。Gal-3与新型冠状病毒感染后的肺部炎症和纤维化反应有关,新型冠状病毒感染的肺巨噬细胞亚群形成由髓系细胞触发受体2(triggering receptor expressed on myeloid cells 2, TREM2)和Gal-3表达控制的促纤维化表型,从而触发肺纤维化[24]。
因此,在肺组织受损后异常修复时,持续的炎症反应诱发肺组织纤维化,Gal-3可能通过在炎症反应中的积极作用参与IPF的发生、发展(图1),具体作用机制还需肺纤维化动物模型及IPF患者的体内外实验研究进一步证实。
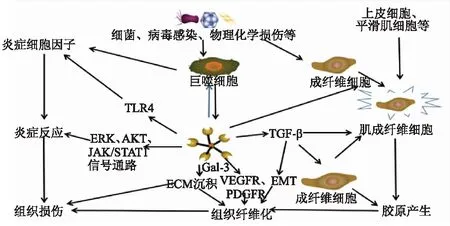
图1 Gal-3在炎症反应和促组织纤维化中的作用及相关分子机制
2.2 Gal-3通过促纤维化作用参与IPF的发生Nishi等[25]首先证明IPF患者BALF中的Gal-3升高,Gal-3诱导THP-1巨噬细胞产生肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)和IL-8;刺激NIH-3T3成纤维细胞诱导迁移和胶原合成。TNF-α是一种纤维原性细胞因子,能够通过巨噬细胞/单核细胞系模型诱导透明质酸-3的产生,可导致细胞外基质异常沉积[8]。Nishi等[25]亦发现患有其他间质性肺病的患者,如隐源性机化性肺炎、急性过敏性肺炎和传染性支气管炎在BALF中Gal-3无明显增加,说明Gal-3表达增加在IPF和与胶原血管疾病相关的间质性肺炎中可能具有特异性。Mackinnon等[4]再次证明IPF患者BALF和血清中Gal-3特异性升高,且在IPF加重期血清Gal-3水平显著升高,提示Gal-3可能是IPF疾病活动和加重的生物学标志物。Gal-3基因敲除小鼠和博莱霉素诱导的小鼠肺纤维化明显减轻,表现为TGF-β诱导的上皮-间质转化(epithelial-mesenchymal transition, EMT)、肌成纤维细胞活化减少和胶原生成减少;Gal-3还降低了β-连环蛋白的磷酸化和核移位[4]。β-连环蛋白通过调节细胞周期蛋白-D1和基质金属蛋白酶7(matrix metalloproteinase 7, MMP7)的表达,与EMT和肺组织重塑有关[26]。在对2 596例弗雷明翰心脏研究参与者进行随访发现,Gal-3浓度升高与肺间质异常和限制性肺功能障碍相关,提示Gal-3在肺纤维化早期有潜在作用[27]。Gal-3还能够交联包括TGF-β受体、血管内皮生长因子(vascular endothelial growth factor, VEGF)受体和血小板衍生生长因子(platelet derived growth factor, PDGF)受体促进纤维化形成[28-29]。TGF-β通过诱导IPF患者中的EMT、肌成纤维细胞活化、细胞外基质生成和肺泡上皮细胞凋亡而促进肺纤维化[30]。PDGF通过其促有丝分裂和趋化作用促进纤维化,还可以与TGF-β协同促进组织纤维化[31]。新型酪氨酸激酶抑制剂尼达尼布就是通过抑制PDGF、VEGF和成纤维细胞衍生生长因子(fibroblast growth factor, FGF)活性,减少胶原沉积和抑制促纤维化基因表达来减轻肺纤维化[32]。Gal-3还可以介导IL-4诱导的巨噬细胞替代激活,IL-4激活的巨噬细胞上调纤维化相关基因,刺激基质生成,进而促进组织纤维化[8]。研究还发现Gal-3是TREM2和分泌性磷蛋白1(secreted phosphorprotein 1, SPP1)协同作用最强的分子之一[33],而TREM2和SPP1是促纤维化巨噬细胞亚群中的纤维化标志物[13]。TLR4与成纤维细胞活化和随后的肺纤维化有关,小鼠模型中TLR4的成纤维细胞特异性缺失诱导肺纤维化显著减少,Gal-3与TLR4的表达有相互促进的作用[21,34]。
综上所述,Gal-3通过与巨噬细胞、成纤维细胞、肌成纤维细胞及一些促纤维化介质相互作用促进肺组织纤维化,参与IPF的发生、发展。
3 Gal-3抑制剂在肺纤维化中的治疗潜力
因Gal-3在炎症反应及促纤维化中的积极作用,使得在纤维化疾病中靶向抑制Gal-3的研究越来越多。目前针对Gal-3抑制剂在IPF治疗中的研究主要为双糖β-硫代半乳糖苷小分子TD139,其是Gal-3碳水化合物结合结构域的新型高亲和力抑制剂[4]。在肺纤维化小鼠的体内外实验中,TD139抑制Gal-3表达,阻断TGF-β诱导的β-连环蛋白的激活,减轻TGF-β及博莱霉素诱导的肺损伤后的纤维化[4,35]。TD139的抗纤维化潜力集中在抑制分泌Gal-3的巨噬细胞的募集和活化,即抑制了局部肌成纤维细胞活化的主要驱动因素[23]。目前已进入临床试验的Gal-3抑制剂是吸入型TD139,在一项临床试验研究中,每天1次以干粉形式吸入给药,持续2周,TD139在肺中高浓度聚集,抑制BALF中巨噬细胞Gal-3表达和与IPF相关的血浆生物标志物的表达,并表现出浓度依赖抑制。该实验还证明吸入TD139在健康受试者和IPF患者中是安全、耐受性良好的,无明显治疗相关副作用,且TD139在肺中的浓度明显高于血液中的浓度[36]。
目前还研发了一种单糖Gal-3抑制剂GB1107,在肺腺癌小鼠模型中经口给药后证明对小鼠肺腺癌有一定的疗效[37]。一项以糖类药物作为内源性生物分子的独特靶向载体或替代物研究发现,双糖药物(TD139替代物)从血液中迅速排出,而单糖药物(GB1107替代物)无排泄迹象。这表明双糖TD139不宜全身用药,而GB1107替代物延长的生物半衰期则表明该药可以全身用药[38]。上述结果进一步说明吸入TD139治疗适用于IPF患者。
4 结语
目前IPF病因及发病机制均不明确,患者预后不良,治疗仍有局限性。Gal-3表达与IPF的发生、发展密切相关,在IPF患者肺泡灌洗液、肺组织匀浆、血清中发现Gal-3水平上升,且在IPF急性加重期显著升高,表明Gal-3有作为IPF及IPF急性加重的生物学标志物的潜力。Gal-3作为一种有效的炎症蛋白,参与炎症反应的启动和放大,是慢性肺损伤纤维化的重要驱动因素。Gal-3还可以通过促进成纤维细胞增殖、肌成纤维细胞活化增殖、EMT、细胞外基质异常沉积等促进肺组织纤维化。即Gal-3通过促进炎症反应、促进肺组织纤维化参与IPF的发生、发展,但具体作用机制仍需更多的肺纤维化动物模型及IPF患者的体内外实验研究进一步证实。对Gal-3抑制剂的临床前实验研究以及对吸入型TD139的临床试验,证明了靶向抑制Gal-3在肺纤维化中的治疗潜力。值得期待的是,吸入TD139与吡非尼酮或尼达尼布联合治疗可能会使IPF患者获得更好的治疗效果,亦需后续试验进行深入探究。
