Investigation of Co-doped Mn oxide catalyst for NH3-SCR activity and SO2/H2O resistance
2022-12-14LIHaixiaJINLeyingZHANGAnchaoSUNZhijunZHANGXinminZHUQifengYANGChangzeZHANGShuaibo
LI Hai-xia ,JIN Le-ying ,ZHANG An-chao ,SUN Zhi-jun ,ZHANG Xin-min ,ZHU Qi-feng ,YANG Chang-ze ,ZHANG Shuai-bo
(School of Mechanical and Power Engineering, Henan Polytechnic University, Jiaozuo 454003, China)
Abstract: A series of Co-doped Mn oxide catalysts with different Co/Mn molar ratios were prepared by co-precipitation method, which was systematically characterized by XRD, SEM, H2-TPR and NH3-TPD etc. Co-doped Mn oxide catalysts are evaluated for NH3-SCR activity and resistance to SO2 and/or H2O, and the Co(1)-MnOx catalyst with Mn/Co molar ratio of 1∶1 performs the best catalytic performance, which achieved higher than 90% NOx conversion in the temperature range of 100-275 °C and possessed better SO2 and H2O resistance. The Co(1)-MnOx catalyst presented a sphere-like structure possessing a relatively large surface area. Doping of cobalt greatly improved the high-valent metal ions and chemisorbed oxygen content of Co(1)-MnOx catalyst surface, and the catalyst possessed abundant active species and acid sites and apparent activation energy of the catalyst was reduced, which makes Co(1)-MnOx a highly effective NH3-SCR catalyst.
Key words: selective catalytic reduction;cobalt-doping;manganese;synergistic effect
Received:2022-03-16;Revised:2022-04-25
*Corresponding author. Tel: 0391-3987546, 0391-3987511, E-mail: lihx@hpu.edu.cn,zhangxm@hpu.edu.cn.
The project was supported by the Key Research Project of Higher Education Institutions in Henan Province (23A470002) and the Innovative Research Team of Henan Polytechnic University (T2020-3).
本文的英文电子版由 Elsevier 出版社在 ScienceDirect 上出版 (http://www.sciencedirect.com/science/journal/18725813)
With the emphasis on environmental protection,people have paid more and more attention to the problem of air pollution. Among the harmful gases,NOx(mainly including NO and NO2) leads to a lot of environmentally harmful effects hazards like acid rain,biochemical haze, depletion of ozone, and global warming effect[1,2]. At present, one of the technologies with the best denitration effect is the NH3selective catalytic reduction technology of NOxby NH3(NH3-SCR)[3,4]. V2O5-WO3/TiO2catalyst has become the most commonly utilized industrial SCR catalyst in the world because of its sulfur resistance and high catalytic activity. But it has problems such as high operating temperature and narrow temperature window (300-400 °C)[5-7]. Therefore, work is underway to provide catalysts that have high catalytic activity, wide operating temperature window.
SCR catalysts can be divided into four main types:precious metal catalysts (Pa, Pt, etc.), metal oxide catalysts (Mn, Co, Ni, Ce, Cr, etc.), and zeolite catalysts(ceramic catalysts), and activated carbon catalysts[8-10].Among them, manganese oxide has become the most developmental low-temperature denitrification catalyst due to its rich valence, acidic sites, and strong redox ability. However, the deteriorated N2selectivity and poor resistance to poisoning SO2and H2O restrict its further industrial application[11]. Therefore, the challenges and opportunities of Mn-based catalysts are to enhance the N2selectivity and resistance to SO2and H2O. The SCR activity of MnOxis MnO2> Mn5O8>Mn2O3> Mn3O4> MnO[12]. The catalytic activity of manganese-based catalyst can be improved by adding other transition metals, such as mixed oxide catalysts produced by mixing manganese and cerium[13], mixing manganese and nickel[14], and mixing manganese and zirconium[15]. In comparison with oxide catalysts containing only one metal, the catalytic activity of composite metal oxide catalysts is greatly improved,and the resistance to water and sulfur is also significantly enhanced due to the increase of the Mn4+proportion. The high valence state of Mn is crucial to the SCR performance for the catalyst.
Cobalt-based materials are well-stocked conductive catalytic feedstocks that can reduce the energy cost required for oxygen evolution reactions by reducing overpotentials[16]. Zhu et al.[17]synthesized Mn-Co binary oxides catalyst of SCR through solvothermal approach. The Mn-Co binary oxides catalyst showed excellent catalytic performance and H2O tolerance. The deNOxefficiency declines in the presence of 1.0 × 10-4SO2from 100% to 58% and recovers gradually to 78%after turning off SO2. Co improves the reducibility,hydrogen uptake and acid base properties of nickel species by increasing the number of strong basic sites[18]. The transition metal cobalt itself is abundant in surface oxygen adsorption, has relatively good redox property and good denitrification performance[19,20].Therefore, we try to prepare cobalt doped manganese metal oxide to obtain a good NH3-SCR catalyst with excellent low temperature activity, wide temperature window and good resistance to SO2/H2O.
As mentioned above, the doping of Co into Mn oxide catalysts could have a positive effect on catalytic performance. In this work, the Co-doped Mn oxide catalysts (Mn: Co = 0.5, 1, 1.5) were prepared by coprecipitation method for NH3-SCR reaction, in which synergistic effect of Mn and Co was explored by XRD,SEM, H2-TPR and NH3-TPD etc. In order to analyze the mechanism of the synergistic effect of manganese and cobalt on reducing denitrification, the denitrification activity and physicochemical properties of the manganese oxide catalyst studied by our research group was cited and compared with that of the novel manganese cobalt composite metal oxides catalyst[15].
1 Experimental
1.1 Materials and methods
Co-doped Mn oxide catalysts were prepared by the co-precipitation method. Co(NO3)2·6H2O and Mn(NO3)2·4H2O with different molar ratio (Mn: Co =0.5, 1, 1.5) were completely dissolved in deionized water, and then the ammonium carbonate was added to the mixture of resultant solution as a precipitate to initiate the coprecipitation reaction. The pH value of mixed solution was adjusted to 9.0-10.0 using NaOH.The obtained solution was stirred at ambient temperature for 2.0 h, and left in air for 12.0 h. The obtained solution was adjusted to a neutral with deionized water, and dried at 110.0 °C for 12.0 h, then,the obtained material was calcined at certain for 4.0 h in muffle furnace. The Co-doped Mn oxide catalysts were denoted by Co(y)-MnOx, andydenoted a molar proportion of Mn and Co. In addition, the single Co and Mn oxide catalysts were prepared by using the same experimental procedure.
1.2 Catalyst characterization
The morphological characterization of catalyst was carried out in a scanning electron microscope(SEM), which was produced by the Japanese company Hitachi. The X-ray diffraction (XRD) test was performed on Bruker D8 Advance in Germany, using CuKα radiation with a scanning range from 10° to 80°,aλof 0.15418 nm, and a speed of 6(°)/min. The N2adsorption and desorption isotherm was the isotherm measured on Quadrasorb EVO with liquid nitrogen N2at -196 °C after degassing in a 300 °C vacuum for 3 h.The H2temperature-programmed reduction (H2-TPR)and temperature-programmed desorption with NH3was carried out using Micromeritics AutoChem II 2920 instrument. Prior to each experiment, the catalysts were subjected to NH3-TPD and H2-TPR, they should be pretreated in helium for 1 h at a temperature of 300 °C with a 20 mL/min flow rate, then cooling to room temperature. The X-ray photoelectron spectroscopy(XPS) test was performed on Semer Escalab 250XI spectrometer. Fourier transform infrared (FT-IR)measurement is performed using the Thermo Fisher Nicolet iS20 spectrometer, Inc.
1.3 NH3-SCR performance measurement
NH3-SCR test experiments were conducted in an adapted fixed-bed reactor with 200 mg sieved to 40-60 mesh catalyst samples at a temperature range of 50-325 °C. The gas hourly space velocity (GHSV) was about 60000 h-1. Reaction gases used: [NO] = 5.0 × 10-4g/mL, [H2O] = 5%, [SO2] =1.0×10-4g/mL, [O2] = 5%,[NH3] = 5.0 ×10-4g/mL (when was used). The NO2,NH3, NO, NO2and N2O at outlet were continuously monitored by the flue gas analyzer. The NOxconversion and N2selectivity were calculated according to the following equations[15].

in those equations, the subscript in and subscript out represented the inlet and outlet gas concentration,respectively.
1.4 NH3-SCR kinetic measurement
Efficient catalysts were those that possess a large turnover frequency (TOF). The TOF of catalytic cycle was an essential property of a catalyst, which does not depend on the rate-determining step or any other single factor, but rather an integrated rate function of the entire cycle. The TOF value was calculated using Equation (6) based on surface active sites amount obtained from H2-TPR characterization results[21]. The activation energy was calculated from the Equation (7)of Arrhenius equation for the temperature range of 75-175 °C.

in the equation,xNOis the conversion rate of NO under the certain SCR reaction temperature;Tis the Kelvin temperature (K);FNOis the NO flow rate (L/min);Ris the gas molar constant (J/(mol·K));Ais the preexponential factor.
2 Results and discussion
2.1 Catalyst crystallographic, morphology and pore structure analysis
2.1.1 XRD analysis
Figure 1(a) presented the XRD patterns of single CoOx, MnOxand Co-doped Mn oxide catalysts prepared at a calcination temperature of 400 °C. There three diffraction peaks of the spinel phase MnOxwere observed, corresponding to the (100), (101) and (102)planes of MnO2(PDF#89-5171), implying (100) facet MnO2was the main form of MnOxprepared by manganese nitrate coprecipitation method. The spectrum of CoOxexhibited seven peaks representing the (111), (220), (311), (400), (422), (511) and (440)planes of Co3O4(PDF#73-1701), indicating the oxide produced from cobalt nitrate using co-precipitation method was Co3O4, (311) was main facet of which. The four diffraction peaks of Co(1)-MnOxand Co(1.5)-MnOxcatalysts at 2θ= 36.8°, 44.8°, 59.5° and 65.3°belonging to CoMn2O4.5, which were attributed to(311), (400), (511) and (440) facet (PDF#32-0297).The lattice parameters of catalysts were calculated by Jade software. The phase composition of Co(1)-MnOxwas CoMn2O4.5, and the lattice parameters wasα=β=γ= 90°,a=b=c= 8.078 Å. The phase composition of Co(1.5)-MnOxwas CoMn2O4.5, and the lattice parameters wasα=β=γ= 90°,a=b=c= 8.075 Å. In contrast, the spectrum of Co(0.5)-MnOxcatalyst exhibited five diffraction peaks, two of which were belonged to CoMn2O4.5and the others were assigned to(Co, Mn)/(Co, Mn)2O4(PDF#18-0408), which proved that the more Co content, the easier it was to form CoMn2O4.5[22]. However, all the formed peaks were relatively weak, indicating that the degree of crystallization was low, and most of the components were amorphous.
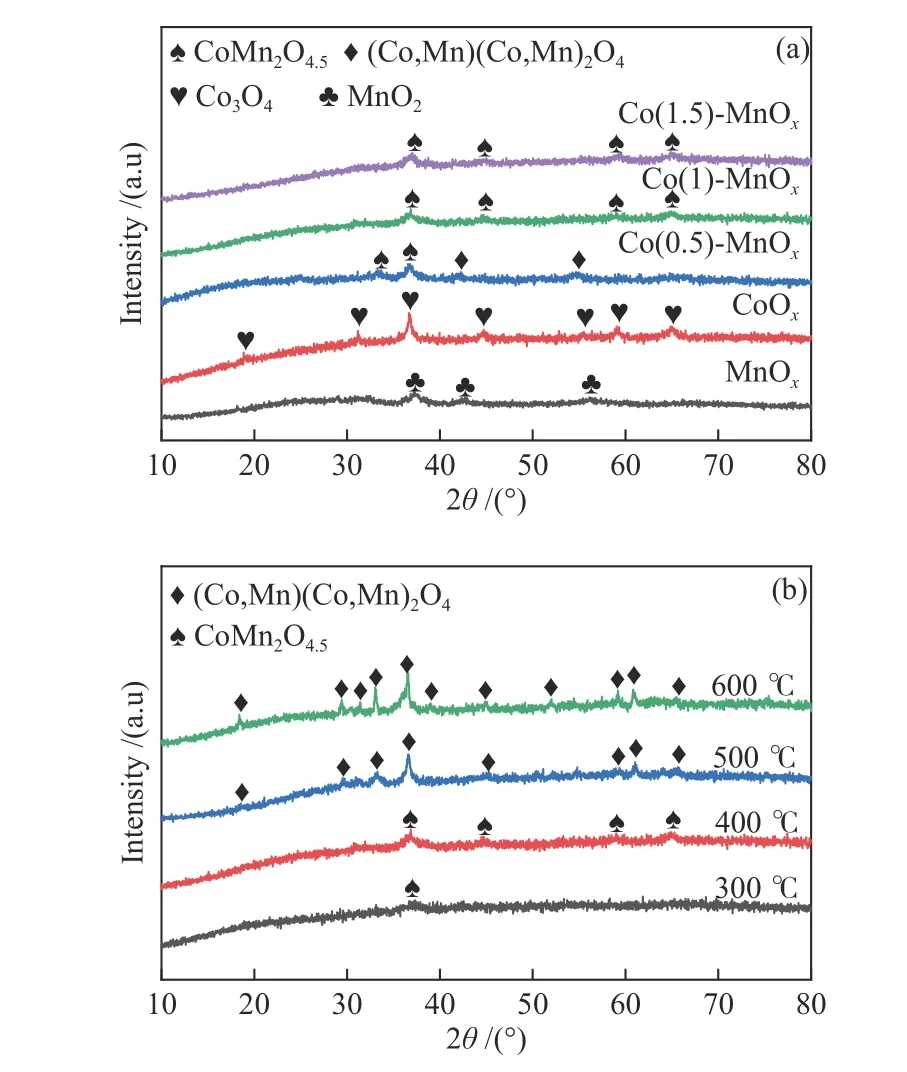
Figure 1 (a) XRD patterns of all the prepared catalysts calcined at 400 °C, (b) XRD patterns of Co(1)-MnOx calcined at 300, 400, 500, 600 °C
The XRD patterns of Co(1)-MnOxcatalysts calcined at different temperatures (300, 400, 500,600 °C) were depicted in Figure 1(b). It can be seen that all the diffraction peaks patterns of Co(1)-MnOxcatalysts calcined at 500 and 600 °C belong to (Co,Mn) (Co, Mn)2O4(PDF#18-0408), and that of 300 °C only exhibited one broad and weak diffraction peak belonging to CoMn2O4.5, proving that 400 °C was the best temperature for formation of CoMn2O4.5.
2.1.2 SEM analysis
All the prepared catalysts calcined at 400 °C were characterized by SEM to study the structure and morphology of the catalysts. (a) and (b) show SEM images of MnOxcatalysts. The spherical structure of the MnOxcatalyst was irregular with relatively rough surface, and many particles were gathered together.Figure 2(a) and (b) showed the external morphology of CoOxcatalyst. It can be observed that CoOxcatalyst showed the morphology of irregular particles. The MnOxand Co(1)-MnOxcatalysts exhibited similar morphological characteristics and the both two were composed of spherical particles[15]. The Co(1)-MnOxcatalyst surface was smoother, and the shape of microsphere structure produced was more regular,while the microsphere structure size of Co-doped Mn oxide catalysts was more uneven than that of MnOxcatalyst. As a result, the Co-doped Mn catalyst particles presented large space and small aggregation, so the surface area and gap were large, and it was favorable for the adsorption and desorption of reacting gas on the catalyst surface, promoting the NH3-SCR reaction.

Figure 2 SEM images of (a), (b) CoOx and (c), (d) Co(1)-MnOx catalysts calcined at 400 °C
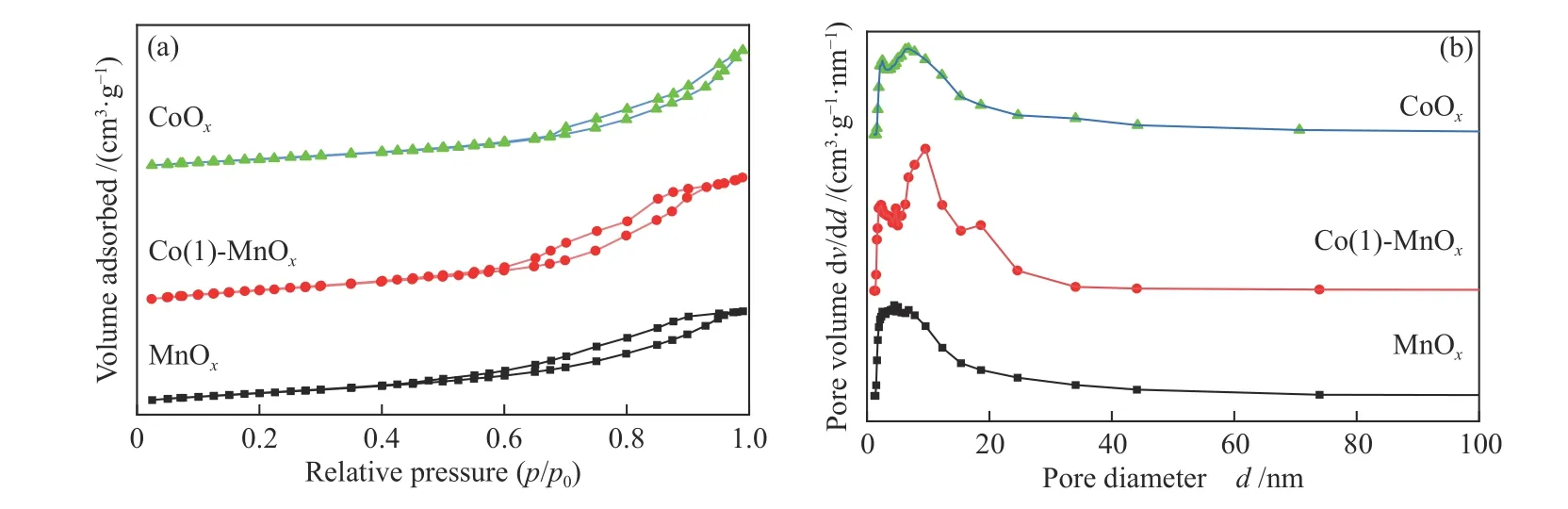
Figure 3 Adsorption-desorption profiles of N2 (a) and distribution of pore size (b) for MnOx, CoOx and Co(1)-MnOx catalysts
2.1.3 N2 adsorption-desorption analysis
The MnOx, CoOx, and Co(1)-MnOxcatalysts calcined at 400 °C were characterized by N2adsorption-desorption, and the obtained isotherm results were shown in Figure 3(a). At lowp/p0, the isotherm presented a less obvious concave downward state. With the increasingp/p0, the MnOx, Co(1)-MnOxand CoOxcatalysts showed hysteresis successively.According to the classification of isotherms and hysteresis loops, it can be seen that N2adsorptiondesorption result of CoOxcatalyst belonged to type IV according to the IUPAC classification, and H3 hysteresis loops[23]. The MnOxand Co(1)-MnOxcatalysts showed a similar change trend, belonging to type IV, H2 hysteresis ring. The H2 hysteresis ring is usually caused by porous adsorbates or uniform particle accumulation pores, and the H3 hysteresis ring was usually caused by layered aggregates, which consistent with the results of SEM characterization.The hysteresis phenomenon was due to the condensation and accumulation of N2in the pressure range. The BET surface area, average pore diameter,and pore volume of catalyst were obtained by mesoporous analysis of the section.
The BET surface area (158 m2/g) of Co(1)-MnOxcatalyst was much larger than that of MnOx(131 m2/g)and CoOx(117 m2/g), as shown in Table 1. The pore volume size obtained was Co(1)-MnOx(0.31 cm3/g) >CoOx(0.29 cm3/g) > MnOx(0.23 cm3/g). The CoOxcatalyst exhibited the largest average pore size, which proved that the doping of MnOxwith Co greatly increased the BET surface area and pore volume. It can be conducive to the formation of catalytic active sites due to the large pore volume, BET surface area and small pores, improving the NH3-SCR activity[24,25].Comparing the catalytic activity results, it was clearly that increasing catalyst pore volume and BET surface area was one of the important links to improve the SCR performance, while pore size was not the key influence.
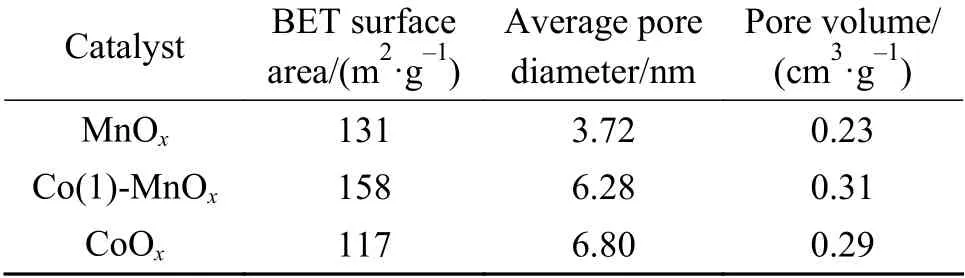
Table 1 MnOx, CoOx and Co(1)-MnOx catalysts structural parameters
2.2 Surface species
2.2.1 H2-TPR
The CoOx, MnOx, and Co(1)-MnOxcatalysts were characterized to study their redox properties by H2-TPR. It can be seen from the XRD pattern that MnOxmostly existed as MnO2. There were two obvious reduction peaks for H2-TPR of MnOxin Figure 4[15].The first one centered at 282 °C results from MnO2→Mn3O4, and the second one centered at 407 °C is attributed to the reduction from Mn3O4→ MnO[2,22].The CoOxspectrum has three obvious reduction peaks centered at 210, 294, and 504 °C, which can be attributed to Co3+to Co2+reduction, Co2+to CoO reduction, and CoO reduction, respectively[26]. The spectrum of the Co-Mn bimetallic oxide Co(1)-MnOxshowed three obvious peaks centered at 217, 303, and 467 °C, first of which belongs to Co3+→ Co2+(and/or MnO2→ Mn2O3), Co2+→ Co ( and/or Mn2O3→Mn3O4), and reduction of CoO process or Mn3O4→MnO, respectively[27]. The low-temperature reduction peak of Co(1)-MnOxshifted to lower temperature, suggesting that the enhancement reducibility of which. Furthermore, the H2consumption of the three catalysts was CoOx(30.55 mmol/g) >Co(1)-MnOx(25.50 mmol/g) > MnOx(17.57 mmol/g).Combining with the catalytic performance, high catalytic activity of the CoOxand Co(1)-MnOxcatalysts at low-temperatures was likely to be ascribed to the surface abundance reducing species, which influenced the catalytic activity significantly.
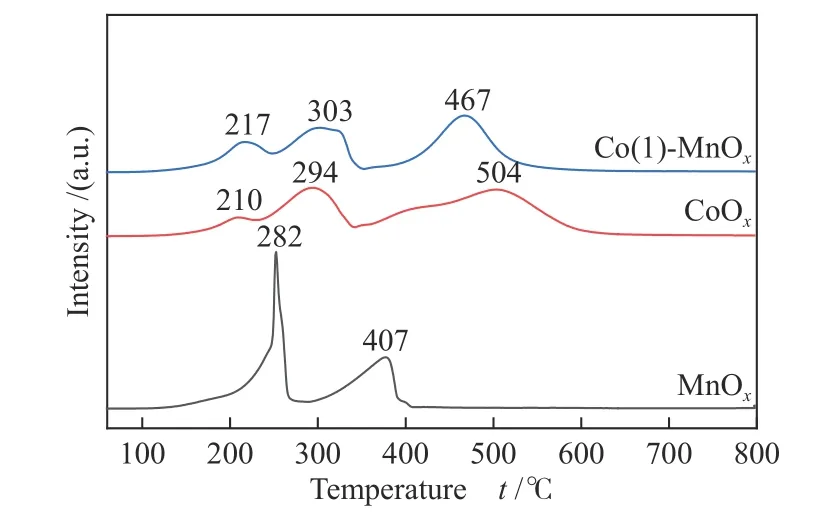
Figure 4 H2-TPR profiles of MnOx, CoOx, and Co(1)-MnOx catalysts
2.2.2 NH3-TPD analysis
NH3-TPD was conducted to obtain the surface acidity of MnOx, CoOx, and Co(1)-MnOxcatalysts(Figure 5). MnOx, CoOx, and Co(1)-MnOxall exhibited three desorption peaks. The three high-temperature peaks of MnOxbelonged to coordinated NH3that bind to the medium-strong acid and strong acid[15]. The desorption peaks at 424 and 486 °C belonged to medium-strong acids, while the desorption peak at 742 °C was formed by NH3adsorption on strong acid sites. For CoOxand Co(1)-MnOxcatalysts, the desorption peak at less than 250 °C belonged to weakly acidicadsorption[28].CoOxand Co(1)-MnOxcatalysts at 254 (258) and 472 °C(492 °C) belonged to NH3adsorption on mediumstrong acid sites. The temperature and area of desorption peak was related to catalyst acid intensity and quantity, respectively. The total amount of NH3desorption was MnOx(17.26 mmol/g) > Co(1)-MnOx(12.52 mmol/g) > CoOx(8.58 mmol/g). The low temperature desorption amount of NH3was Co(1)-MnOx(2.32 mmol/g) > CoOx(2.02 mmol/g), indicating that the doping of Co increased catalyst surface weak acidic sites and enhanced NH3absorption and desorption capacity at low temperature. Corresponding to the low-temperature catalytic activity, it was further proved that the low-temperature catalytic activity can be effectively improved by added weak acid sites[29].Thus, Co-doped MnOxcatalysts possessed more acid sites, especially medium-strong acid: Co(1)-MnOx(10.2 mmol/g) > CoOx(6.56 mmol/g). Comparing NH3-SCR activity of MnOx, CoOxand Co(1)-MnOxcatalysts, the high catalytic performance achieved by MnOxand Co(1)-MnOxcatalysts at medium and high temperatures were likely to be attributed to abundant acidic sites, which indicated that improving the catalyst apparent acidity was a significant factor to improve the catalytic performance in high and medium temperature NH3-SCR[22,30].
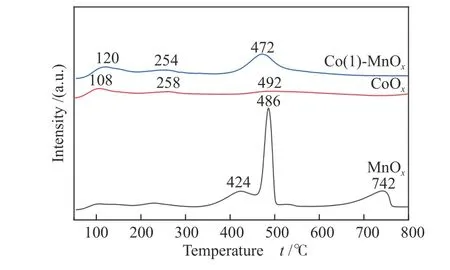
Figure 5 NH3-TPD profiles of MnOx, CoOx, and Co(1)-MnOx catalysts
2.2.3 XPS analysis
In order to research the element composition and atomic valence composition of catalyst surface, the Co(1)-MnOxand MnOxcatalysts calcined at 400 °C were characterized by XPS. The surface atom contents were calculated, and the results were shown in Table 2.The proportion of O and Mn on Co(1)-MnOxcatalyst surface was much higher than that in MnOxcatalyst,which was consistent with the XRD characterization results. This showed there was a change in the structure of the MnOxcatalyst by the doping of Co, and more Mn and O atoms were formed on the surface.

Table 2 Surface element concentrations and relative atomic concentrations of Co(1)-MnOx and MnOx catalysts
It was observed from Figure 6(a) that the XPS spectra of Mn 2pwas divided into two characteristic peaks, one peak for the Mn 2p1/2and another peak for the Mn 2p3/2. Each characteristic peak contains three binding energy peaks. The peak of Mn 2p3/2at 641.08-641.32 eV belongs to Mn2+, the peak at 642.32-642.38 eV belongs to Mn3+, and the peak at 643.83-644.12 eV belongs to Mn4+[31]. In comparison with manganese oxide catalysts, the Mn2+of Co-doped bimetallic oxide catalysts decreased, while the Mn3+and Mn4+increased significantly[15]. The proportion of Mn4+was Co(1)-MnOx(37.34%) > MnOx(33.14%),while the proportion of Mn3+(39.49%) of Co(1)-MnOxcatalyst was much larger than that of MnOxcatalyst(35.06%). As reported, the redox-oxidation capacity can be improved by increasing the content of Mn4+[32].According to H2-TPR results, the Co(1)-MnOxcatalyst showed stronger reducibility than MnOxcatalyst, which proved that the increasing Mn4+improved the oxidation-reduction ability. In a low-temperature catalytic environment, the proportion of high-valent metal ions was an important factor affecting the catalytic activity, the more high-valent metal ions the better catalytic performance[33]. This was consistent with the low-temperature NH3-SCR catalytic efficiency of Co(1)-MnOxand MnOxcatalysts. In addition, Mn4+was beneficial for increasing the proportion of surface adsorbed oxygen.

Figure 6 (a) Mn 2p, (b) Co 2p and (c) O 1s XPS profiles of Co(1)-MnOx and MnOx catalysts
The Co 2pXPS profile was shown in Figure 6(b).There were two spin orbitals of Co 2p3/2and Co 2p1/2,which can be fitted into three peaks. The peaks of Co 2pat 780.28 and 795.33 eV were attributed to Co3+,the peaks at 781.98 and 797.08 eV belonged to Co2+,and the peaks at 786.83 and 803.13 eV belonged to satellite peaks[34,35]. The relative content of Co3+can be obtained from the area of the spectra, Table 2 showed that Co3+/Co of Co(1)-MnOxcatalyst was 33.26%. Co3+was the highest valence state of metallic Co. Combined with the NH3-SCR performance, it can be found that Co(1)-MnOxcatalyst showed the best catalytic performance, which proved that Co3+distributed was a significant factor.
The O 1sXPS profile was shown in Figure 6(c). It can be seen that there were obvious binding peaks in the two ranges of 529.61-530.08 eV and 530.98-531.42 eV[32-36]. The peak with lower binding energy was related to chemically adsorbed oxygen(Oα), and high binding energy one was attributed to lattice oxygen (Oβ)[37]. Surface chemically adsorbed oxygen was the most active oxygen due to its relatively high mobility, which was more conducive to the occurrence of oxidation reactions[38]. The proportion of surface chemisorbed oxygen (Oα/(Oα+Oβ)) of Co(1)-MnOxand MnOxcatalysts were 58.20% and 49.70%,respectively, indicating that the more abundant Oαin Co(1)-MnOxcatalyst surface, which was beneficial to the oxidation reaction of NO to NO2[15,37].
2.3 SCR performance
Figure 7(a) illustrated the de NOxperformance of MnOx, CoOx, and Co(y)-MnOxcatalysts calcined at 400 °C for NH3-SCR reaction, and GHSV = 60000 h-1.The CoOxcatalyst presented a lower activity temperature range than MnOxcatalyst, which achieved a high NOxconversion at 75-250 °C, while that for MnOxcatalyst was 125-275 °C. As shown in Figure 7(b), the highest N2selectivity for Co(0.5)-MnOx, Co(1)-MnOx, and Co(1.5)-MnOxwas higher after 150 °C, especially MnOx. The experimental results showed that Co-doped MnOxcatalysts can improve the N2selectivity of single metal oxide. The CoOxcatalyst exhibited the highest N2O selectivity, while MnOxcatalyst was the lowest(Figure 7(c)), which corresponds to their N2selectivity at less than 250 °C. More N2O produced during the catalytic process will reduce the N2selectivity of the catalysts. It can be seen from Figure 7(d) that the NH3conversion of CoOxrate to reach 100% at the lowest temperature of 125 °C, while the NH3conversion of Co(1)-MnOxcatalyst reach 100% at the highest temperature of 225 °C. Figure 7(e) shows that the NO2selectivity of all the catalysts was relatively low and stable, indicating that the catalysts produce less NO2during the catalytic reduction process. The catalytic activity of Co(0.5)-MnOxcatalyst was closest to that of MnOxcatalyst, while the overall catalytic activity was improved. The Co(1)-MnOxcatalyst achieved 90% NOxconversion rate at 100 °C, and maintained 100% in the temperature range of 100-250 °C. The bimetallic oxide catalysts produced by Co-doped MnOxcatalyst can compensate for the poor catalytic effect of MnOxcatalyst at low-temperatures as well as CoOxcatalyst at high temperatures, and greatly broaden the temperature range for high catalytic performance[15].
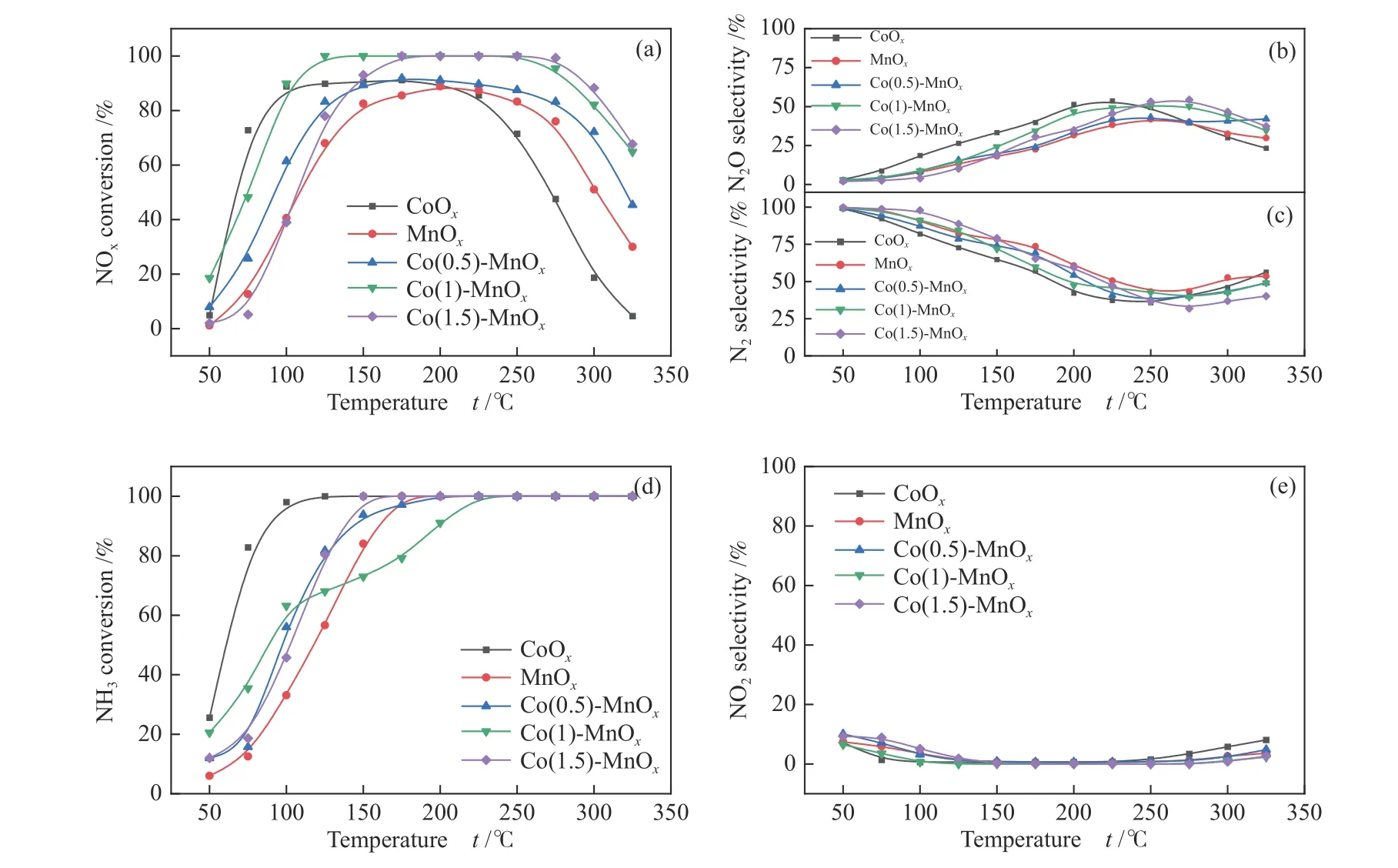
Figure 7 (a) NOx conversion, (b) N2O selectivity, (c) N2 selectivity, (d) NH3 conversion and(e) NO2 selectivity of MnOx, CoOx, and Co(y)-MnOx
2.4 Catalytic performances
The catalyst reaction rate was calculated according to formula (6) and all the kinetic parameters obtained from the temperature range of 50-150 °C[39].As shown in Figure 8, the TOF value change trend of the three catalysts was roughly consistent with the changing trend of the NOxconversion. The TOF value increased gradually as the increasing temperature, the highest TOF (3.76×10-3s-1) of Co(1)-MnOxcatalyst occurred at 125 °C. According to the H2-TPR analysis result, owing to the excellent redox properties of CoOxcatalyst, more NH3would be absorb and reacted with NO during the initial catalytic reaction process, higher catalytic efficiency at low temperatures was reached.As the catalytic temperature increased, MnOxgradually reached the equal TOF value as Co(1)-MnOxcatalyst[40,41]. Due to the abundance of active sites on the Co(1)-MnOxcatalyst surface, it showed a higher catalyst reaction rate. According to the catalytic results,it was proved that the higher TOF value was, the more favorable the catalyst reacted with NH3and NOx[41,42].
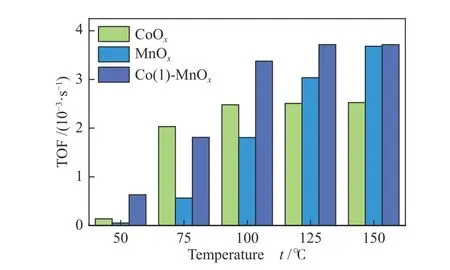
Figure 8 TOF values of MnOx, CoOx, and Co(1)-MnOx catalysts in 50-150 °C
The apparent activation energy (Ea) of MnOx,CoOx, and Co(1)-MnOxcatalysts can be obtained from the slope of Arrhenius plots shown in Figure 9, which were (52.07 ± 2.57), (27.75 ± 1.48), and (21.55 ± 1.76)kJ/mol, respectively. The apparent activation energy of the Co(1)-MnOxcatalyst was significantly reduced, the reaction barrier was significantly reduced[43], which was consistent with catalytic activity.Eavalue of MnOxwas above 40.00 kJ/mol, which indicated that the NH3-SCR process over MnOxcatalyst at low temperature was controlled by chemical reaction control[5]. TheEaof Co(1)-MnOxcatalysts prepared by co-precipitation method decreased significantly compared with other methods reported in the literatures, such as hydrothermal method and solvothermal method[8,43]. For a chemical reaction, the lower the activation energy,the faster the reaction rate. Therefore, The SCR reaction over the Co(1)-MnOxcatalysts prepared by coprecipitation method is faster.
2.5 Catalytic performance of calcination at different temperatures
The NOxconversion and N2selectivity of Co(1)-MnOxcatalyst prepared with different calcination temperatures (300-600 °C) were presented in Figure 10.Among them, the Co(1)-MnOxcatalyst calcined at 400 °C showed the highest NOxconversion and N2selectivity in a relatively wide temperature range. The catalytic performance of catalyst calcined at 300, 500 and 600 °C was significantly worse compared with the catalytic activity at 400 °C, which proved that the calcination temperature played an important role in the preparation of catalyst. For Co-doped MnOxcatalysts,the best catalytic activity was obtained for the Co(1)-MnOxcatalyst calcined at 400 °C. Figure 10(c) showed that the Co(1)-MnOxcatalyst calcined at 500 °C possessed the highest N2O selectivity, corresponding to its lowest N2selectivity. Figure 10(e) presented that the catalyst calcined at 400 °C the lowest and most stable NO2selectivity, which indicated that 400 °C was the most favorable temperature for Co(1)-MnOxcatalytic activity.
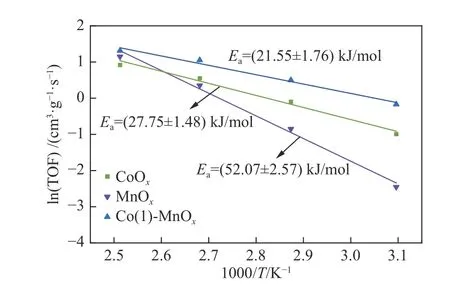
Figure 9 Arrhenius plots of MnOx, CoOx and Co(1)-MnOx catalysts
2.6 Resistance to SO2 and H2O of Co(1)-MnOx catalyst
As shown in Figure 11, 100 mg/L SO2(and/or 5%H2O) was introduced to research resistance to SO2and H2O of Co(1)-MnOxcatalyst at 150 °C. In the first reaction hour, NOxconversion of Co(1)-MnOxalmost maintained at 100% without 100 mg/L SO2. The NOxconversion significantly reduced from 100% to 86%within 0.5 h, and then gradually increases and remained at around 91% within the subsequent 5 h as the introduction of 100 mg/L SO2. The NOxconversion rate returned to 95% in 1 h after SO2was stopped without any additional treatment. Although the NOxconversion of 100% cannot be restored, the Co(1)-MnOxcatalyst exhibited much excellent SO2resistance.The poisoning effect of SO2on the catalyst was significantly exacerbated by the presence of water, 5%H2O were further introduced into the reaction, NOxconversion was reduced from 100% to 87%, and restored to 92% in 1 h after SO2and H2O were stopped.The results show that the Co(1)-MnOxcatalyst has a certain degree of resistance to sulfur and water.
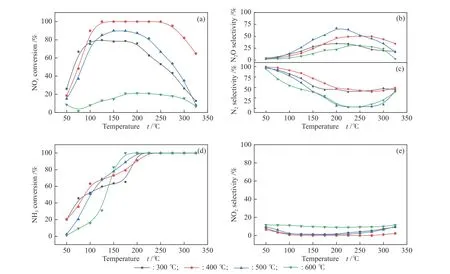
Figure 10 (a) NOx conversion, (b) N2O selectivity, (c) N2 selectivity, (d) NH3 conversion and (e) NO2 selectivity of Co(1)-MnOx calcined at different temperatures
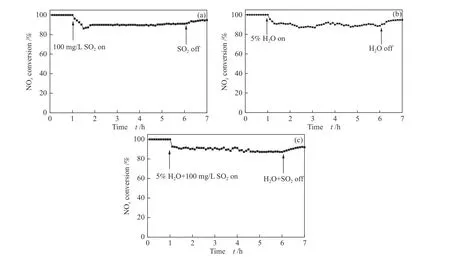
Figure 11 Effect of H2O and SO2 at 150 °C on NH3-SCR performance of Co(1)-MnOx catalyst: (a) SO2 resistance test,(b) H2O resistance test,(c) H2O + SO2 resistance test
The Co(1)-MnOxand the Co(1)-MnOxafter the sulfur resistance test at 150 °C were characterized by FT-IR, and the changes of surface groups were observed. As shown in Figure 12, four bands were detected at 1117, 1396, 1630, and 3360 cm-1for the two catalysts, respectively. The bands at 1630 and 3360 cm-1were attributed toδHOH(absorption band of HOH bending mode) and stretching of hydroxyl groups. The band at 1396 cm-1was due to the symmetric stretching vibration of N[44,45]. While a new band was found in the surface of 150 °C-SO2-Poisoning Co(1)-MnOx, the band at 1117 cm-1corresponded to the sulfate species. SO2gas molecule would react with the catalyst surface active sites and sulfate formed, and the catalytic activity was inhibited by the occupation of sulfate species. Competing adsorption of SO2on the active sites of the catalyst surface with each reaction gas, so the catalytic activity was improved as the introduction of SO2was stopped.The effect on catalyst activity of sulphate species already deposited on the catalyst was irreversible[46].

Figure 12 FT-IR spectra of fresh and 150 °C-SO2-Poisoning Co(1)-MnOx catalysts
As reported[47], the reversible deactivation caused by H2O due to competitive adsorption with the reactants, while the irreversible deactivation may be owing to the formation of surface hydroxyl groups. The NOxconversion of Co(1)-MnOxrecovered quickly when H2O was stopped, and it can be inferred that the inhibition of catalyst activity by H2O was largely reversible. The excellent SO2and H2O resistance of Co(1)-MnOxwas due to its abundant active sites.
2.7 Synergistic effect of Mn and Co in NH3-SCR reaction
Different surface structures, redox, and acidity further affect the SCR catalytic activity of the catalysts.The variation of metal molar ratios in catalysts will affect morphology and structure, changing the specific surface area and the composition of the physical phases on the surface. The larger the pore volume and specific surface area, the more uniform adsorption sites can be distributed. Increasing NO and NH3adsorption at the active sites can also provide better diffusion for gas generated after the reaction[48-50]. Calcining at different temperatures was to make the catalyst react violently between molecules to form a crystal structure at a relatively high temperature, and provided more adsorption centers. It can be obtained by SEM, XRD,and BET characterization that the catalyst calcined at 400 °C with a molar ratio of 1:1 formed an obvious spherical structure with a higher degree of crystallization and a Co-Mn bimetallic oxide phase CoMn2O4.5, Co(1)-MnOxcatalyst showed the largest pore volume and specific surface area, that was conducive to the formation of catalytic centers and can disperse active components, adsorbing more reaction gases for reaction and increasing the SCR activity.
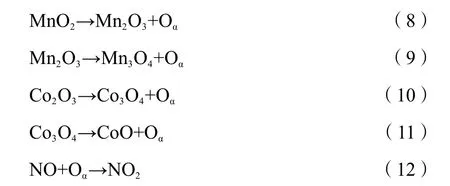
The synergistic effect of Co and Mn promoted the catalytic activity of Co(1)-MnOxcatalyst. The reaction mechanism of the selective catalytic reduction process was shown in Figure 13. Among the MnOx, CoOx, and Co(1)-MnOxcatalysts, CoOxshowed the strongest redox properties, MnOxsurface contained abundant acid sites. Co(1)-MnOxcombines the redox and acidity of two single-metal catalysts. The surface of the catalyst contains abundant active and acid sites, and this greatly improved the adsorption capacity. The major active oxides on Co(1)-MnOxcatalyst surface were MnO2and Co3O4. The intense interaction between Mn and Co promoted the electron transfer between metal ions. As shown in Formula (9)-(12), a large amount of lattice oxygen with high fluidity was generated in the catalyst, which promoted the cycle of oxygen transfer on catalyst surface: O2(gas) ↔ O2(ads)↔ O2-(ads) ↔ 2O-(ads) ↔2O2-(lattice), which further promoted NO oxidation[51]. It was also because of the strong interaction between oxides that it was easy to generate NO2and N2O, which reduced the N2selectivity.
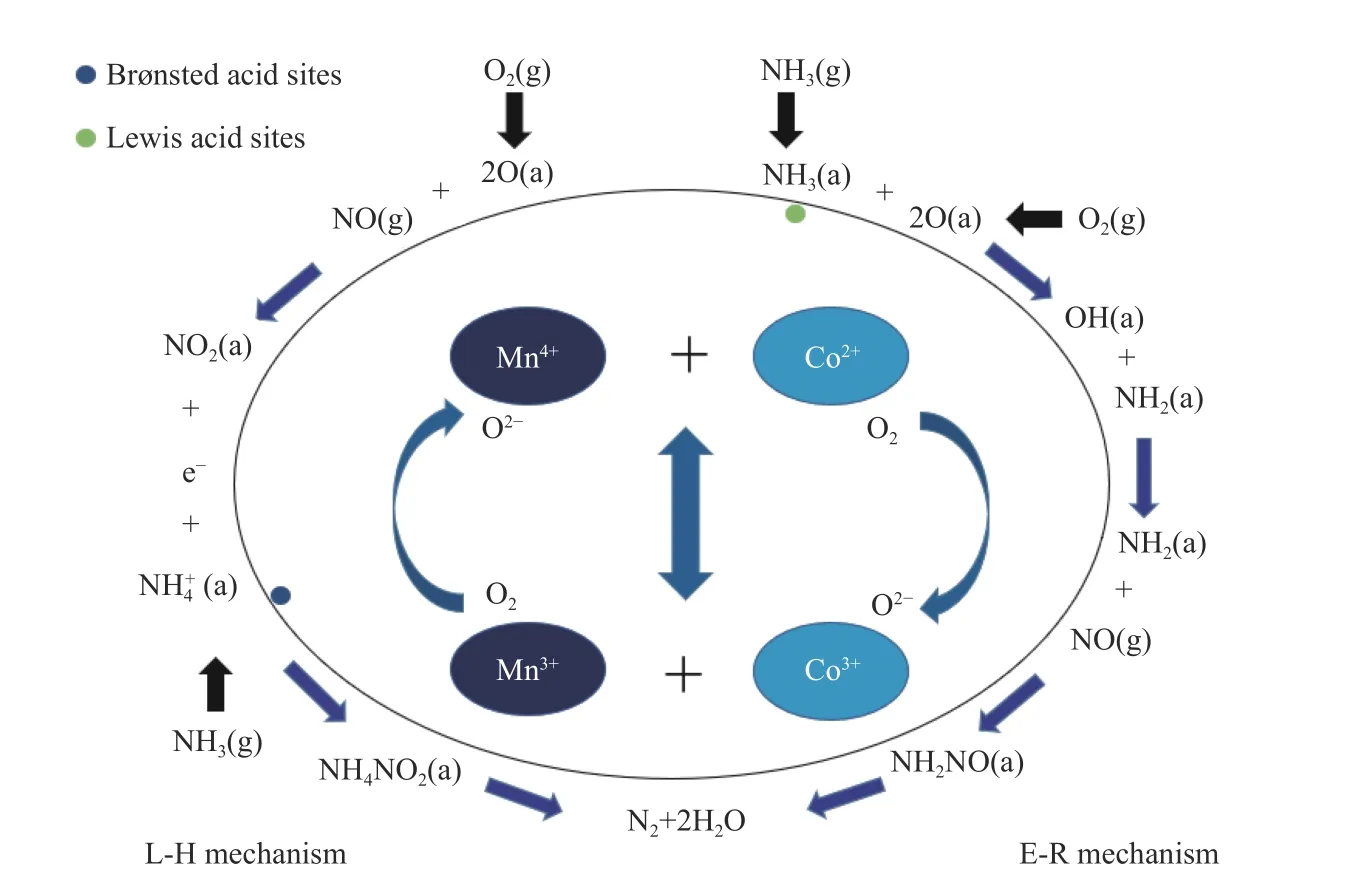
Figure 13 NH3-SCR reaction mechanism on Co(1)-MnOx catalyst surface
According to previous studies, Mn-based catalysts generally showed two reaction mechanisms: Eley-Ridea (E-R) and Langmuir-Hinshelwood (L-H)mechanism[47]. The L-H mechanism required that the reacted molecules should be adsorbed on catalyst surface. The E-R mechanism only required one of molecules participating in the reaction should be adsorbed on catalyst surface[52,53]. A coordinated NH3was formed by adsorption onto the Lewis acid site on the catalyst surface. The adsorbed NH3was oxidized to generate adsorbed NH2and OH. Then NH2continued to react with NO gas molecules to form the transition product NH2NO, finally, it readily decomposed to H2O and N2. This process belonged to E-R reaction mechanism. O2was adsorbed on the catalyst surface to form adsorbed oxygen, the adsorbed oxygen reacted with NO to generate NO2, and NH3was adsorbed on the Brønsted acid site to form adsorbed NNO2reacted with adsorbed Nto generate transition product NH4NO2, and NH4NO2was decomposed into N2and H2O. This process belonged to the L-H reaction mechanism[52,54,55].
3 Conclusions
The Co(1)-MnOxwith Co/Mn molar ratio of 1:1 exhibited the best NH3-SCR Porous bimetallic catalytic performance, reaching 90% NOxconversion at 100 °C,and maintaining 100% NOxconversion in the whole temperature region (125-250 °C). In addition, the Co(1)-MnOxcatalyst exhibited excellent the smallest apparentEaand SO2and/or H2O resistance, and Co(1)-MnOx. The comparison proved that the Co-doped Mn oxide catalyst produced by the co-precipitation method showed superior performance.
After Co doping with Mn, the catalyst structure and form were changed, forming a clear spherical structure, and the Co(1)-MnOxcatalyst specific surface area was significantly increased. The Co(1)-MnOxcatalyst was mainly composed of CoMn2O4.5according to XRD results. The analysis of XPS, NH3-TPD, and H2-TPR showed the surface Mn4+, Co3+, and chemisorption oxygen content of the Co(1)-MnOxcatalyst was the most abundant, which increased the acid sites and catalyst active species. The doping of Mn with Co relatively increased the redox capacity and acidity, improving NO and NH3adsorption ability of the catalyst surface, and the NH3-SCR catalytic activity was further improved.
