HPLC梯度洗脱-多波长切换法同时测定固肾丸中4种成分含量*
2022-12-09李鹂贺蓓蓓黄良永杜士明
李鹂,贺蓓蓓,黄良永,杜士明,3
[1.十堰市太和医院(湖北医药学院附属医院)药学部,十堰 442000;2.湖北医药学院药学院,十堰 442000;3.武当特色中药研究湖北省重点实验室(湖北医药学院),十堰 442000]
固肾丸是我院自制的中药制剂(批准文号:鄂药制字Z20210292)。该制剂在我院临床使用了近30年,已被证明具有很好的疗效。其主要成分为淫羊藿、白芍、制何首乌、女贞子、熟地、菟丝子、当归等17味中药。具有养血健脾、温肾固本、补阴助阳、调节内分泌平衡的作用。用于肝肾亏虚所致的四肢无力,腰膝酸软;女子垂体功能障碍、宫冷不孕、男子肾虚不育等症。该制剂标准收载于《湖北省医疗机构制剂规范》(2011版),标准中仅有淫羊藿苷的含量测定项[1]。而组方中女贞子、白芍、制何首乌、淫羊藿均为固肾丸的君药。为了提升该制剂的质量标准,全面反映君药的有效成分,保证制剂的有效性,本研究建立一种新的高效液相色谱(HPLC)梯度洗脱-多波长切换法,同时对淫羊藿中主要成分淫羊藿苷、白芍的主要成分芍药苷、制何首乌的主要成分二苯乙烯苷、女贞子的主要成分红景天苷的含量进行测定,并对含量测定方法进行验证,确定4种测定成分的含量限度,为修订和提高该制剂的质量标准提供科学依据,也为下一步该制剂的HPLC指纹图谱研究提供方法学参考。
1 仪器与材料
1.1仪器 ThermoFisher Ultimate 3000 HPLC(美国赛默飞公司,LPG-3400泵、WPS-3000SL analyt进样器、TCC-3000柱温箱、VWD-3400四波长检测器);变色龙色谱工作站v6.8中文版;色谱柱:OMNIBond HPLC column Orca ( C18,4.6 mm×250 mm,5μm );科盟牌超声清洗机(KM-36C,40 kHz,0~250 W可调,广州科洁盟科技公司);SHIMADZU AUW120D电子分析天平(感量:0.01 mg,日本岛津公司);GWA-UN Pure Water system(北京普析通用仪器公司)等。
1.2药品与试剂 红景天苷对照品(批号:200605,含量:98%)、芍药苷对照品(批号:110736-202044,含量:96.8%)、二苯乙烯苷对照品(批号:110844-201109,含量:94.7%)、淫羊藿苷对照品(批号:110737-200415,110737-200413,含量:100%)均购自中国食品药品检定研究院。中药饮片:熟地(批号:20210401)、盐菟丝子(批号:20210301)白芍(批号:20210401)、川芎(批号:20210401)、醋延胡索(批号:20210301)、砂仁(批号:20200701)、党参(批号:20210401)、当归(批号:20210301)、醋香附(批号:20210101)、枸杞子(批号:20210301)、覆盆子(批号:20201001)、女贞子(批号:20210301)、乌附片(批号:20200601)、佛手(批号:20200801)、制何首乌(批号:20210401)、淫羊藿(批号:20200801)由湖北清大中药饮片有限公司生产;黄芪(批号:20210401)由湖北药昇中药科技有限公司生产。固肾丸3批次(批号:20210806,批号:20210506,批号:20210320,规格:10.5 g×45丸/盒)均由我院制剂室生产。乙腈(德国默克公司)为色谱纯;磷酸、乙醇、甲醇等试剂均为分析纯;水为自制超纯水。
2 方法与结果
2.1固肾丸的制备 处方:淫羊藿420 g、白芍225 g、何首乌280 g、女贞子280 g、熟地420 g、当归225 g、川芎140 g、醋延胡索225 g、砂仁84 g、党参252 g、黄芪420 g、覆盆子225 g、盐菟丝子280 g、枸杞子225 g、乌附片140 g、佛手225 g、香附225 g。
制备:按处方取中药材,洗净后烘干,粉碎成细粉,过筛(80目),钴60辐照灭菌,与蜂蜜混匀(1:1),制成大蜜丸,即得。
2.2色谱条件 以OMNIBond HPLC Orca C18(4.6 mm×250 mm,5 μm)为色谱柱;以0.1%磷酸溶液(A)-乙腈(B)为流动相,按表1进行梯度洗脱;按表2中规定时间-波长程序检测,波长分别为:220,230,320,270 nm;柱温:30 ℃;流速1.0 mL·min-1,理论塔板数以芍药苷的峰计算应不低于8000。

表1 梯度洗脱程序 Tab.1 Program of gradient elution

表2 时间-检测波长程序 Tab.2 Program of time-wavelength
2.3溶液的制备
2.3.1供试品溶液的制备 分别取3批次(批号:20210806,20210506,20210320)固肾丸,各10丸,剪碎成细粒状,混匀,取3.0 g,精密称定,分别置于100 mL具塞锥形瓶中,精密加入50%乙醇35 mL,称定质量,热水浴中将样品充分溶散后,超声处理(功率250 W,频率40 kHz)60 min,放冷至室温,擦干锥形瓶外水分后再称定质量,用50%乙醇补足减失的质量,摇匀,静置,取上清液滤过,取续滤液,即得。
2.3.2对照品溶液的制备 芍药苷、二苯乙烯苷、淫羊藿苷对照品溶液配制:分别精密称取芍药苷、二苯乙烯苷、淫羊藿苷对照品11.9,10.87,10.73 mg,各置10 mL量瓶中,避光操作,加甲醇溶解并稀释至刻度,摇匀,制成单一对照品贮备液 (1.1519,1.0294,1.073 mg·mL-1),冷藏,备用。
红景天苷对照品溶液配制:取红景天苷对照品13.64 mg,精密称定,置25 mL量瓶中加甲醇溶解并稀释至刻度,摇匀,制成浓度为0.5347 mg·mL-1的对照品储备液,冷藏,备用。
2.3.3阴性样品溶液 按处方比例分别称取缺女贞子、白芍、制何首乌、淫羊藿的饮片粉末共约1.5 g,蜂蜜1.5 g,分别置于100 mL具塞锥形瓶中,按“2.3.1”项下方法操作,即得4种阴性样品溶液。
2.4系统适应性实验 分别精密吸取红景天苷对照品溶液 0.5 mL、芍药苷对照品溶液0.5 mL、二苯乙烯苷对照品溶液0.35 mL、淫羊藿苷对照品溶液0.4 mL,置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即成质量浓度分别为26.735,57.595,36.029,42.92 μg·mL-1的混合对照品溶液。精密吸取供试品溶液(批号:20210806)、混合对照品溶液及4种阴性样品溶液,按“2.2”项下色谱条件进样,记录色谱图,结果见图1。可知,红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷的保留时间分别是17.20,28.00,35.16,51.27 min;理论塔板数均不低于8000;各待测成分的色谱峰与相邻峰的分离度均大于1.5;阴性样对照品色谱图中均无干扰成分。

1:红景天苷;2.芍药苷;3.二苯乙烯苷;4.淫羊藿苷;A.混合对照品溶液;B.供试品溶液;C.缺女贞子阴性样品;D.缺白芍阴性样品;E.缺何首乌阴性样品;F.缺淫羊藿阴性样品。图1 固肾丸中4种成分的色谱图 1.Salidroside;2.paeoniflorin;3.2,3,5,4'-tetrahydroxy stilbene-2-O-β-D-glucoside;4.Icariin;A.mixed refernce;;B.test solution;C.negative sample without privet;D.negative sample without white peony;E.negative sample without polygonum multiflorum;F.negative sample without herba epimedii.Fig.1 Typical chromatograms of 4 components in Gushen wan
2.5线性关系的考察 分别精密吸取“2.3.2”项下对照品储备溶液适量,置同一量瓶中,加甲醇稀释成5组混合对照品溶液,浓度分别为红景天苷(267.350,133.675,26.735,5.347,2.674 μg·mL-1),芍药苷(575.95,287.975,57.595,11.519,5.7595 μg·mL-1),二苯乙烯苷(360.29,180.145,36.029,7.205 8,3.602 9 μg·mL-1),淫羊藿苷(429.2,214.6,42.92,8.584,4.292 μg·mL-1)。分别按“2.2”项下的色谱条件进样测定,并记录各待测成分峰面积。分别以4个待测成分的质量浓度为横坐标(X,μg·mL-1)、相应峰面积为纵坐标(Y)进行线性回归,得回归方程和线性范围,结果见表3。

表3 固肾丸中4种成分的线性关系考察结果 Tab.3 Linear relationships of four constituents in Gushen wan
2.6重复性实验 取同一批固肾丸(批号:20210806)10丸,剪碎,混匀,平行称取6份,每份3.0 g,精密称定,按照“2.3.1”项下方法制成6份供试品溶液,然后按照“2.2”项下的色谱条件,精密吸取各供试品溶液连续进样2次,记录各待测成分峰面积平均值。结果,红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷峰面积的RSD(n=6)分别为1.91%,1.21%,0.91%,0.79%,表明该方法重复性较好。
2.7精密度实验 精密吸取“2.5”项下第三组混合对照品溶液(中间浓度),连续进样6次,记录各待测成分峰面积。结果红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷峰面积RSD(n=6)分别为1.37%,1.77%,1.27%,1.39%,表明仪器精密度良好。
2.8稳定性实验 取“2.3.1”项下供试品溶液(批号:20210806)适量,于室温下避光放置,在分别经过1,2,4,8,12,24 h后,精密吸取该供试品溶液按“2.2”项下的色谱条件进样测定,记录各待测成分峰面积。结果,红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷的峰面积RSD(n=6)分别为0.99%,0.76%,0.43%,0.67%,表明供试品溶液在室温下避光放置24 h内稳定性良好。
2.9加样回收率实验 将已知含量的同一批固肾丸(批号:20210806)10丸,剪碎,混匀,称取6份,每份约1.5 g,精密称定,分别置于100 mL具塞锥形瓶瓶中。向6个锥形瓶中各精密加入“2.3.3”项下红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷对照品储备液液0.80,0.95,0.60,0.75 mL,用50%乙醇补充至35.0 mL,按照“2.3.1”项下方法制成供试品溶液,然后按照“2.2”项下的色谱条件进样测定,记录各待测成分峰面积,将峰面积代入线性方程计算各成分的含量及加样回收率。结果红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷的平均加样回收率分别为 101.41%,98.47%,101.32%,101.55%,RSD(n=6)均小于2%,表明该方法准确度好,见表4。
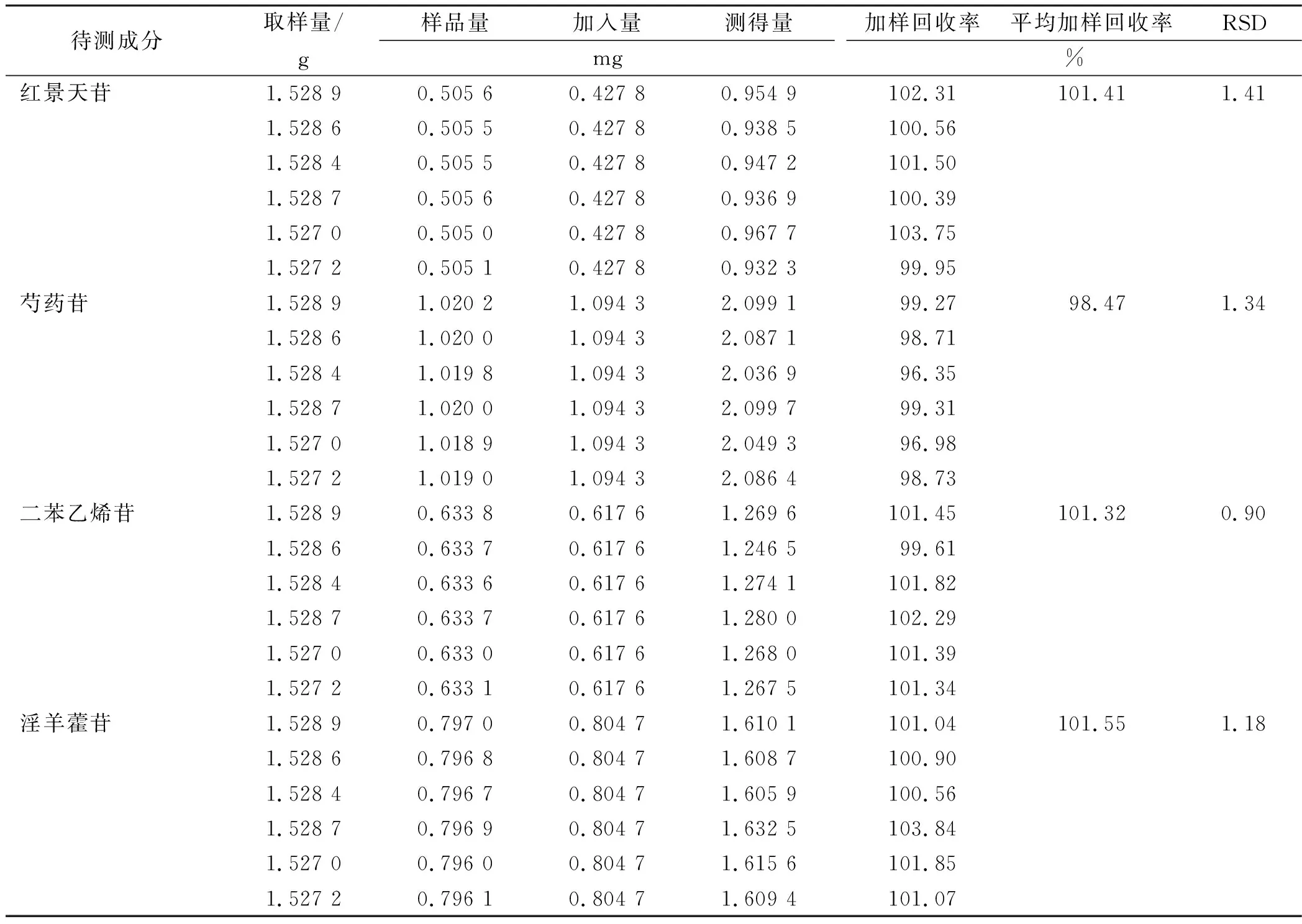
表4 固肾丸中4种成分的加样回收实验结果 Tab.4 Recovery rates of 4 components in Gushen wann=6
2.10样品测定 取“2.3.1” 项下制备好的3批供试品溶液,按照“2.2”项下的色谱条件进样测定,记录各待测成分峰面积,将峰面积代入线性方程计算样品中各成分的含量。以干燥品计算,3批固肾丸(批号分别为20210806,20210506,20210320)的水分含量分别是7.3%,5.0%,6.1%);结果3批供试品溶液中红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷的平均含量见表5。

表5 3批固肾丸中4种成分的含量测定结果 Tab.5 Results of content determination in Gushen wanmg·g-1,n=3
3 讨论
3.1色谱条件的选择 本研究采用HPLC梯度洗脱、多波长切换法同时测定固肾丸中4种成分的含量。前期流动相考察了乙腈-0.1%磷酸溶液和乙腈-0.05%磷酸溶液等不同流动相体系,经过反复实验发现以乙腈-0.1%磷酸溶液为流动相按表2进行梯度洗脱时,色谱图的基线平稳,各色谱峰能较为完全分离,各测定成分峰型较好。检测波长选择,依据《中华人民共和国药典》一部中[2]女贞子、白芍、制何首乌、淫羊藿药材含量测定项下,红景天苷、芍药苷、二苯乙烯苷、淫羊藿苷的最佳检测波长分别为220,230,320,270 nm,因此本研究选择采用多波长切换法,结合色谱图中的各测定成分保留时间采用时间-波长程序控制最佳检测波长,提高各测定成分的检测灵敏度。
3.2提取方法的选择 本研究前期考察了超声提取[3-8]、加热回流提取[3-4,9]、SPE提取[10-14],提取时间均为60 min,结果发现,采用上述方法提取时,样品中的测定成分含量无明显差异,对固肾丸中测定成分提取效率基本相同,考虑到操作的简便性,故采用超声提取法(40 kHz,250 W);采用单因素优化法,对超声提取的超声时间(30,60,90 min)进行考察,发现超声60,90 min时提取效率差异较小,但均优于超声30 min,故采用60 min作为超声时间。提取溶剂的选择,《中国药典》中[2]女贞子、白芍、制何首乌、淫羊藿药材的含量测定均采用50%乙醇提取,本研究也对提取溶剂单因素优化,采用40%,50%,70%,100%乙醇及相同浓度的甲醇超声比较,结果显示50%乙醇对测定的4种成分提取效率均最高,故本研究最终采用50%乙醇为溶剂超声提取(40 kHz,250 W)60 min作为提取方法。
3.3含量测定结果分析 本研究采用HPLC梯度洗脱多波长切换法同时测定了固肾丸中的4种成分含量,方法学验证结果表明该法操作简便、重复性好、准确度高。测定结果表明,3批样品中4种待测成分的平均含量从高到低依次为芍药苷(0.65~0.72 mg·g-1)、淫羊藿苷(0.35~0.56 mg·g-1)、二苯乙烯苷(0.30~0.56 mg·g-1)、红景天苷(0.19~0.36)mg·g-1,各批间4种成分的含量有明显差异,这可能与饮片的质量差异有关,为保证制剂成品的质量,中药饮片的来源应统一,质量应严格把关;结果也表明多成分测定对控制固肾丸质量的必要性。根据测定结果,按测定成分的平均含量的80%作为含量限度指标,本品中芍药苷、淫羊藿苷、二苯乙烯苷、红景天苷含量按干燥品计算须≥0.56,0.36,0.35,0.20 mg·g-1。
