辣黄白通便复方颗粒剂处方与生产工艺研究
2022-12-05伊博文李梦薇任佳伟
伊博文, 李梦薇, 郑 蕊, 任佳伟
(1.中国中医科学院西苑医院,北京 100091;2.北京中医药大学中药学院,北京 102488;3.华北电力大学医院,北京 102206)
质量源于设计(QbD)是一种基于风险评估,强调对产品特性系统了解的基础之上,对产品从源头设计到生产制造过程进行系统和可靠的控制,以提高产品的质量[1]。2004年FDA定义“过程分析技术”[2-3],2009年人用药品注册技术国际协调会议(ICH)在ICH Q8(R2)中提出该理论[4],2013年FDA要求制药企业必须采用该理念用于药品的质量控制[5],2017年我国正式成为ICH成员。至此,QbD理念已经逐渐被制药界达成在控制药品质量和减少产品上市后风险的共识。
辣黄白通便复方由辣木叶、黄芪、白术3味药材组成,具有益气健脾、增强机体免疫力、抗氧化作用,用于脾虚气弱的特定人群[6-8],对便秘有较好的疗效[9-10]。提取工艺研究,该方日生药量精制后出膏率达19.9%,不宜制成普通口服片剂和胶囊剂,为方便服用,适应大规模生产,颗粒剂是一种合适的剂型。
本研究采用QbD理念中的确定关键质量属性(CQAs),风险评估和实验设计(DoE)方法,研究辣黄白通便复方颗粒剂处方与生产工艺,以期为该制剂质量控制提供参考。
1 材料
1.1 仪器 电子天平(型号Sartorius BSA2202、CPA225D,德国赛多利斯公司);电子调温电热套(型号DZTW);旋转蒸发仪(型号RE-52AA);水浴锅(型号XMTD-6000);混合机(型号SYH-20,南京科迪信机械设备有限公司);湿法制粒机(型号G10,深圳市信宜特科技有限公司);真空干燥箱(型号DZ-2BC,天津市泰斯特仪器有限公司);整粒机(型号DGN-II,上海优译柯机械科技有限公司);高效液相色谱仪(型号LC-16)、紫外检测器(型号SPD-16)(日本岛津公司)。
1.2 试剂与药物 异槲皮苷对照品(111809-201403,纯度92.9%,中国食品药品检定研究院);紫云英苷对照品(批号Y27J7H9634,纯度99.5%,上海源叶生物科技有限公司)。辣木叶(云南天佑科技开发有限公司);黄芪(批号1611020,北京太洋树康中药饮片厂);白术(批号1609003,北京太洋树康中药饮片厂),均经北京中医药大学中药鉴定系张贵君教授鉴定为正品。食品添加剂规格乳糖、甘露醇、糊精、麦芽糊精、可溶性淀粉。乙腈为色谱纯(批号164790,美国Fisher公司);甲醇(批号20170209)、磷酸(批号20160111)为分析纯(北京化工厂);水为娃哈哈纯净水(批号201102GQ07547,杭州娃哈哈集团有限公司)。实验设计、数据统计分析均由JMP 11.0.0平台实现(SAS Institute Inc.)
2 方法与结果
2.1 处方优化
2.1.1 浸膏粉制备 按处方量分别称取辣木叶、黄芪、白术,10倍量水煎煮2次,每次2 h,滤过,合并滤液,80%乙醇进行醇沉处理,上清液减压浓缩干燥成干膏粉,即得。
2.1.2 颗粒剂制备 称取过40目筛后浸膏粉和辅料(乳糖、甘露醇、糊精、麦芽糊精、可溶性淀粉)各7.5 g,混合后过40目筛3次,使其混合均匀。将干浸膏粉和辅料放入蒸发皿内,滴加80%乙醇并用手不断搓捏,混匀至“手捏成团、压之即散”,制软材,在10目筛上制粒,置于烘箱中,在60 ℃下干燥,20目整粒,即得。
2.1.3 辅料筛选 CQAs是指与产品有效性和安全性相关的质量属性[11],参照2020年版《中国药典》四部通则0104,确定颗粒剂CQAs为成型率、溶化性、水分、装量差异。虽然处方优化时会对颗粒进行干燥,测定水分没有意义,但颗粒吸湿性会影响其包装保存后含水量,而流动性关系到后期制剂生产时的装量差异,因此,确定处方CQAs为成型率(>85%)、流动性(豪斯比<1.11)、溶化性(5 min内在70~80 ℃中热水应全部溶解)、吸湿性(越小越好)。本实验分别设置成型率比重占25分,豪斯比比重占25分,溶化性比重占20分(完全溶解)或10分(不溶解),吸湿性比重占30分。
2.1.3.1 成型率 参考文献[12]报道,制备好的颗粒称定质量,先过1号筛(10目)再过5号筛(80目),收集能通过1号筛但不能通过5号筛者称定质量,计算成型率,公式为成型率=(过筛后颗粒质量/过筛前颗粒质量)×100%,得分=(25/最大成型率)×成型率。
2.1.3.2 流动性 参考文献[13]报道。
堆密度:将过筛后颗粒置于干燥量筒中,轻轻振动,读出其近刻度处的体积数V,以过筛后的质量为M,堆密度=M/V。
振实密度:将过筛后颗粒置于干燥量筒中,举起3 mm,每分钟轻叩250次,共3 min,读出其近刻度处的体积数V,以过筛后的质量为M,振实密度=M/V。
豪斯比:豪斯比=振实密度/堆密度,得分=(25×最小豪斯比)/豪斯比。
2.1.3.3 溶化性 参考文献[12]报道,取颗粒约2 g,加40 mL热水溶解,搅拌,观察溶液状态。
2.1.3.4 吸湿性 参考文献[12]报道,取一定量颗粒,置于30 ℃烘箱中恒重48 h,移到预先饱和好的相对湿度75%的玻璃干燥器中,在已恒重的扁称量瓶底部放入厚约2 mm的颗粒,准确称定质量,置于干燥器中(扁称量瓶打开)24 h,再称定质量,计算吸湿率,公式为吸湿率=[(吸湿后颗粒质量-吸湿前颗粒质量)/吸湿前颗粒质量]×100%,得分=(30×最小吸湿率)/吸湿率。
2.1.3.5 综合评分 参考文献[14-15]报道,对“2.1.3.1”至“2.1.3.4”项下结果进行汇总,结果见表1。由此可知,糊精溶化性较差;麦芽糊精吸湿性较大;甘露醇成型率、流动性(豪斯比超过1.11)不理想;可溶性淀粉成型率较低,吸湿性大;乳糖各CQAs良好,综合评分最高。因此,选择乳糖作为辅料。

表1 辅料优化结果
2.1.4 乳糖用量 按浸膏粉-乳糖 1∶1、1∶2、2∶1、1∶1.5比例混合均匀,加入适量80%乙醇适量,制软材,在1号筛(10目)上制粒,湿颗粒置于烘箱中,在60 ℃下真空减压干燥,即得,其成型率、豪斯比见表2。由此可知,尽管不同比例浸膏粉-乳糖下上述2个CQAs均符合要求,但为1∶1时颗粒成型率最高,流动性最好。因此,选择1∶1作为浸膏粉与乳糖比例。

表2 乳糖用量考察结果
2.1.5 验证试验 按上述优化处方制备3批样品,发现其外观均匀,成型率分别为95.5%、93.8%、96.6%,流动性良好(豪斯比分别为1.07、1.06、1.05),颗粒均可在5 min内完全溶于热水中,48 h吸湿率分别为9.5%、10.8%、11.9%。
2.2 生产工艺优化
2.2.1 整体风险评估 依据2020年版《中国药典》四部0104颗粒剂项下有关规定和本品质量标准,首先确定颗粒成型率、流动性、含水量、指标成分含量作为CQAs(由于颗粒溶化性主要与处方组成相关,与生产工艺基本无关,故暂不考虑该CQA),具体见表3(不能在每个工艺步骤中都测定所有CQAs, 以避免过度测定而影响成本和效率)。

表3 生产工艺优化过程中需要重点关注的CQAs
然后,建立与其密切相关的每个生产工艺步骤中间体产出物料CQAs,从而控制终产品颗粒剂质量,实现QbD理念中过程和终点全程质量控制的目的,包括输入物料的关键物料属性和工艺参数,具体见表4。

表4 工艺步骤中产生出物料CQAs和高风险工艺变量评估
2.2.2 原辅料混合工艺开发
2.2.2.1 变量评估 可能影响原辅料混合均匀度的输入物料属性包括复方提取物粒度分布和提取物流动性,以及辅料粒度分布、流动性、松密度,在处方优化时已对其进行控制,均符合颗粒剂要求,故不列入高风险工艺变量;工艺参数包括混合仪器的混合器填充水平、混合桨转速和混合时间,由于两者参数(50%~70%;只有高、低档,通常选择低档300 r/min)相对固定,故也未被列入高风险变量。表4已经确定了混合均匀度为该工艺步骤中产出物料的CQAs,确定混合时间为高风险工艺变量,故将研究混合时间对混合均匀度的影响与该变量的控制。
2.2.2.2 混合时间对混合均匀度的影响 以指标成分含量均匀度[16]为考查指标,采用单因素试验考察混合时间(5~15 min,100袋规模,在混合机上、中、下各5个不同位置取样)对混合均匀度的影响,结果见表5。由此可知,混合时间为10~15 min时,异槲皮苷、紫云英苷含量均匀度RSD均小于3%,符合制剂混合均匀度要求。

表5 原辅料混合设计和混合均匀度测定结果
2.2.3 湿法制粒工艺开发
2.2.3.1 工艺变量评估 可能影响湿法制粒步骤成型率的输入物料属性包括原辅料混合均匀度、润湿剂用量,在原辅料混合工艺步骤中发现原辅料混合10~15 min时,即可确保指标成分含量混合均匀度RSD<3.0%,由于在处方工艺优化时已经确定润湿剂为80%乙醇,用量为20%~30%,故未被列入高风险的工艺变量。该步骤中工艺参数包括混合仪器混合桨转速和制粒时间,由于仪器转速只包括高、低两档,通常选择低档300 r/min,故也未被列入高风险工艺变量。表4中成型率为该工艺步骤中产出物料CQAs,确定制粒时间为高风险工艺变量,故将研究制粒时间对成型率的影响与该变量的控制。
2.2.3.2 制粒时间对成型率的影响 采用单因素试验考察制粒时间(3~9 min,100袋规模,取样10次进行测定)对成型率的影响,结果见表6。由此可知,制粒时间为6~9 min时,成型率>90%,RSD更小,符合制粒要求。
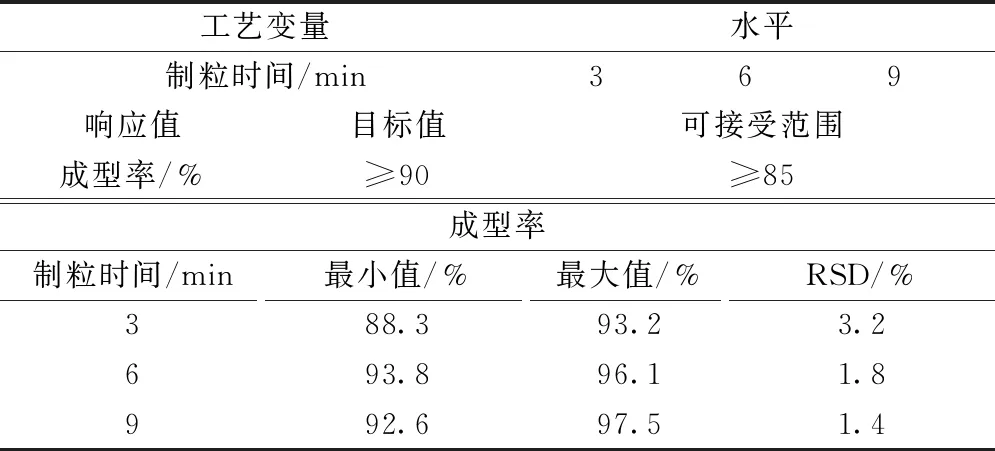
表6 湿法制粒实验设计和成型率测定结果
2.2.4 颗粒干燥工艺开发 基于表4风险评估可知,LOD为CQAs,干燥温度、干燥时间、物料厚度为高风险工艺变量,筛选DoE,评估上述3个关键因素对LOD的影响。采用三因素三水平设计,即因素A (干燥温度40、50、60 ℃)、因素B (干燥时间20、40、60 min) 、因素C (物料厚度0.5、1.0、1.5 cm),LOD可接受范围设定为1.0%~4.0% (由于还有后续工艺步骤,故需要控制该CQAs在更低的水平,才能保证最终成品其数值在8.0%以下)。在干燥颗粒不同位置(上、中、下)取样,共15个样品,计算平均LOD,筛选DoE,结果见表7。
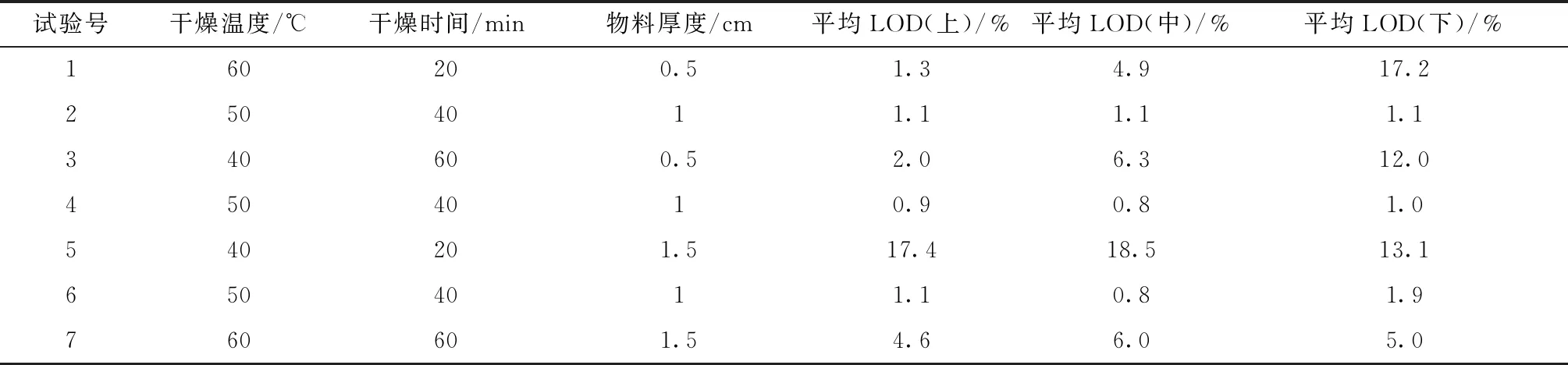
表7 干燥实验设计结果
方差分析见表8,可知3个模型P<0.01,具有高度显著性;R2、校正R2均大于0.95,表明模型拟合度良好;因素C的P<0.01,因素A在LOD(上)、LOD(中)模型中的P<0.01,表明两者具有极显著影响,并且存在交互作用;因素B的P>0.05,表明无显著影响。

表8 方差分析
预测刻画器(图1)显示,干燥步骤的最佳工艺变量组合为干燥温度48 ℃,物料厚度0.99 cm,而干燥时间不是显著因素,故选中间值(40 min)。DS图(图1)给出了干燥工艺环节中2个显著因素的设计空间(白色区域),其中因素A可在40~60 ℃范围内变化,因素C可在0.77~1.5 cm范围内变化,厚度过小时LOD会低于1%,但过大时所需温度更高。另外,可在采用设计空间内的干燥温度和物料厚度均可使干燥后颗粒LOD在4%以内。
2.2.5 颗粒整粒工艺开发
2.2.5.1 工艺变量评估 该步骤中工艺参数包括整粒机速度、筛网类型、筛网目数、整粒环境温湿度,由于所用整粒机的速度不可调节,整粒机筛网只有尼龙筛网和不锈钢筛网(主要根据物料制粒难度而搭配固定筛网类型),故未被列入高风险工艺变量。工厂的温湿度一般会在生产过程中进行监控,同时在后续工艺步骤中进行吸附等温线研究,以明确不会影响物料质量属性者,故也未被列入高风险变量。表4确定成型率为CQAs,整粒筛网目数为高风险工艺变量,故将研究整粒筛网目数对成型率的影响与该变量的控制。
2.2.5.2 整粒筛网目数对成型率的影响 根据仪器设备的实际情况设计3个水平(20、24、30目筛整粒),结果见表9。由此可知,整粒筛网目数在20~30目时成型率均大于90%,符合颗粒剂要求。

表9 整粒工艺设计与结果
2.6 临界相对湿度测定 参考文献[12,17]报道,按上述优化处方和生产工艺制备颗粒剂,取约1 g至干燥恒重并编号的称量瓶中,共9份,置于不同相对湿度的密闭干燥器中,打开称量瓶盖,置于25 ℃恒温箱中48 h,精密称定质量,计算增重,结果见表10。再以吸湿率为纵坐标,相对湿度为横坐标绘制吸湿曲线,2条直线相交点对应的横坐标值即为临界相对湿度,结果见图2,可知该数值约为65%,即制粒、分装、贮存过程中环境温度控制在25 ℃时,相对湿度必须控制在65%以下,可保证环境湿度不会对颗粒制备产生明显影响。

表10 不同相对湿度下颗粒吸湿率
3 讨论与结论
中药制剂的研究主要包括处方研究和生产工艺参数研究,目前发表的文献主要是聚焦于应用QbD理念进行处方研究[18-20], 但生产工艺参数研究和控制也同样是非常重要的,尤其是在确保不同批次间产品的质量稳定性方面。还有一些研究聚集于某一个制剂工艺环节[21-22],但这并不能真正实现对产品质量的整体控制。本研究参考FDA公开的药物QbD研究的案例[23],将QbD理念中的CQAs、风险评估、DoE引入辣黄白通便复方颗粒剂处方和生产工艺参数的设计与优化中,以期实现从设计、中间工艺步骤到终产品的全程质量控制,从而保证产品的稳定、均一、可控。
本实验首先确定了CQAs,采用单因素试验优化辅料种类和用量,发现乳糖在各方面的性质较优越,而且具有良好的冲溶性和抗湿性,它与浸膏粉比例为1∶1时,颗粒容易制粒,成型率、流动性好,易于溶化,吸湿性小。再建立QbD研究过程:(1)确定CQAs为成型率、含水量、流动性、指标成分含量;(2)通过风险评估确定颗粒生产时每个工艺步骤中需要重点关注的CQAs;(3)基于需要重点关注的CQAs,确定中间体产出物料的该参数;(4)确定每个工艺步骤中高风险工艺变量(包括中间体输入物料关键质量属性和关键工艺参数;(5)以中间体产出物料的CQAs为指标,应用单因素和DoE设计空间方法,确定高风险工艺变量的最优值和可接受的范围,以最大程度降低在生产过程中未知因素的潜在风险,从过程控制确保最终颗粒剂产品质量和批次间的稳定性。临界相对湿度(CRH)是药物吸湿率增加的临界值,可根据它作为确定生产过程中的环境湿度的重要参考,本实验测得该参数约为65%,较易控制,即辣黄白通便复方颗粒剂的生产、包装、存贮环境的相对湿度应控制在65%以下,才能保证其顺利制备。
综上所述,本实验基于QbD理念研究了辣黄白通便复方颗粒剂处方组成和生产工艺参数的控制范围,特色之处在于将该理念中的CQAs、风险评估、DoE应用到各个工艺步骤中,并通过风险评估将三者关联到一起,再结合被鉴定的每个工艺步骤中高风险工艺变量,采用单因素或DoE研究其控制范围和最优值,可达到从源头设计、到过程控制和终产品全程质控的目的。
