依匹哌唑的合成工艺研究
2022-11-30彭子祥王瑶琼刘进兵
彭子祥, 刘 丹, 王瑶琼, 刘进兵
(邵阳学院 食品与化学工程学院,湖南 邵阳 422099)
依匹哌唑(Chart 1)是由日本大冢制药和丹麦灵北制药共同研发的治疗精神分裂症药物,美国FDA于2015年7月10日批准其上市,北京康立生医药技术开发有限公司于2016年11月获得依匹哌唑片批件(受理号:JXHL1000032)。依匹哌唑的化学名为:7-[4-(4-(苯并[b]噻吩-4-基)-哌嗪-1-基)丁氧基]-1H喹啉-2-酮,属于5-HT/DA受体调节剂,既可用于治疗成人精神分裂症,也可与抗抑郁药联合用于治疗成人重度抑郁。相对于阿立哌唑,依匹哌唑的疗效及耐受性更好,并且可降低患者静坐不能、不安和失眠等不良反应的发生率[1-4]。
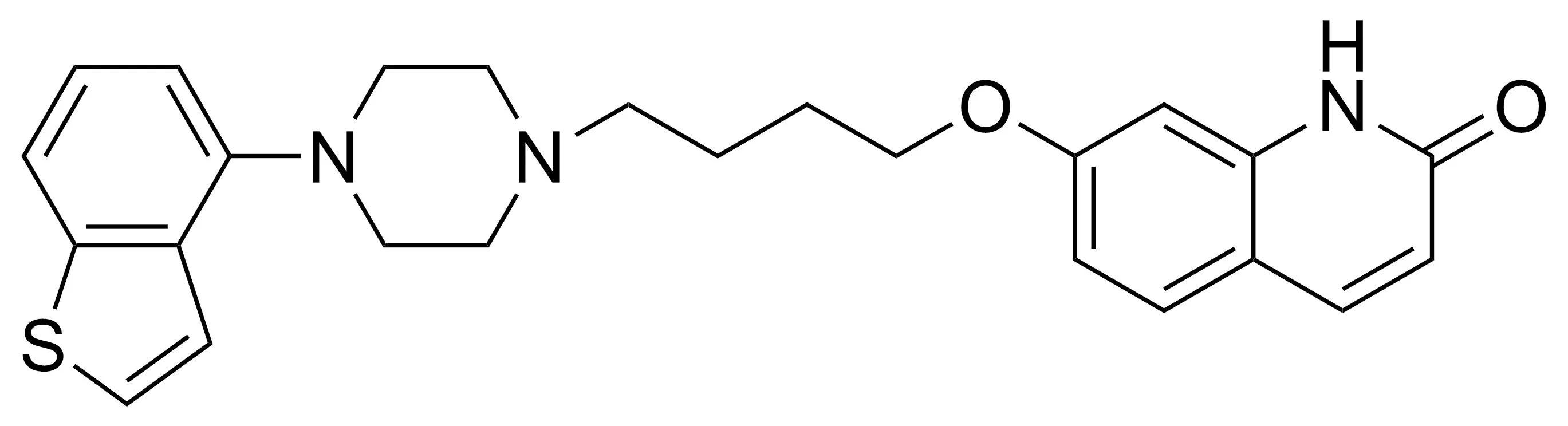
Chart 1
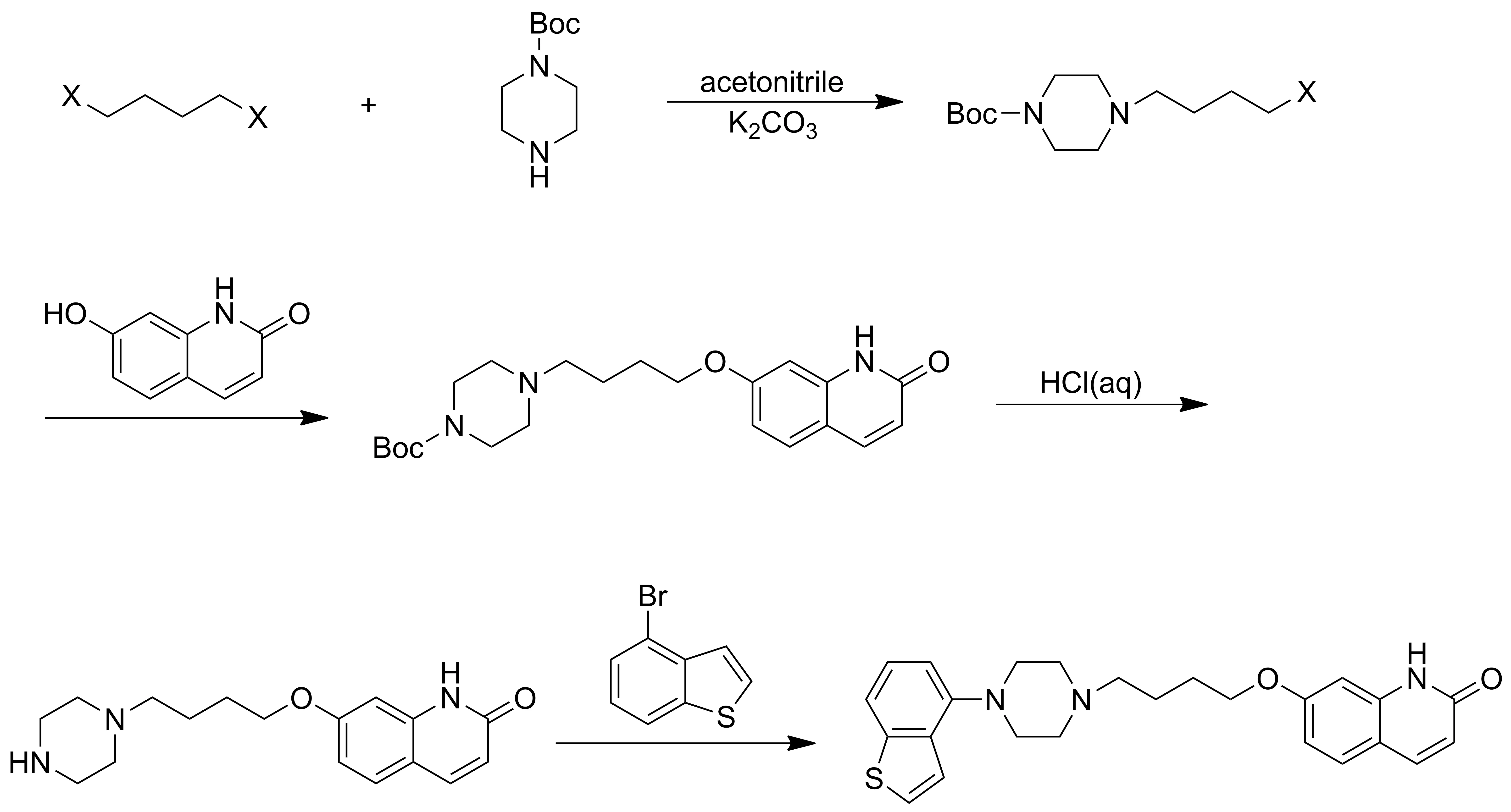
Scheme 1
Scheme 2
关于依匹哌唑的合成方法,已有较多文献报道。Jagtap等[5]将Boc保护的哌嗪与1,4-二取代丁烷反应,取代1,4-二取代丁烷中的一个取代基,得到含有Boc保护哌嗪的丁烷衍生物,将所得的衍生物再和7-羟基喹啉酮反应,得到Boc保护的7-(4-(哌嗪-1-基)丁氧)喹啉-2-(1H)酮,经脱保护后得到7-(4-(哌嗪-1-基)丁氧)喹啉-2-(1H)酮,再与4-溴苯并噻吩反应得到依匹哌唑。该法第一步反应收率不高,且1,4-二取代丁烷两端有同时被哌嗪取代的可能性(Scheme 1)。

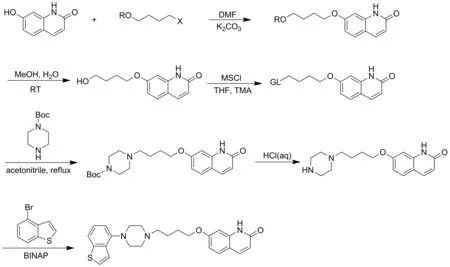
Scheme 3
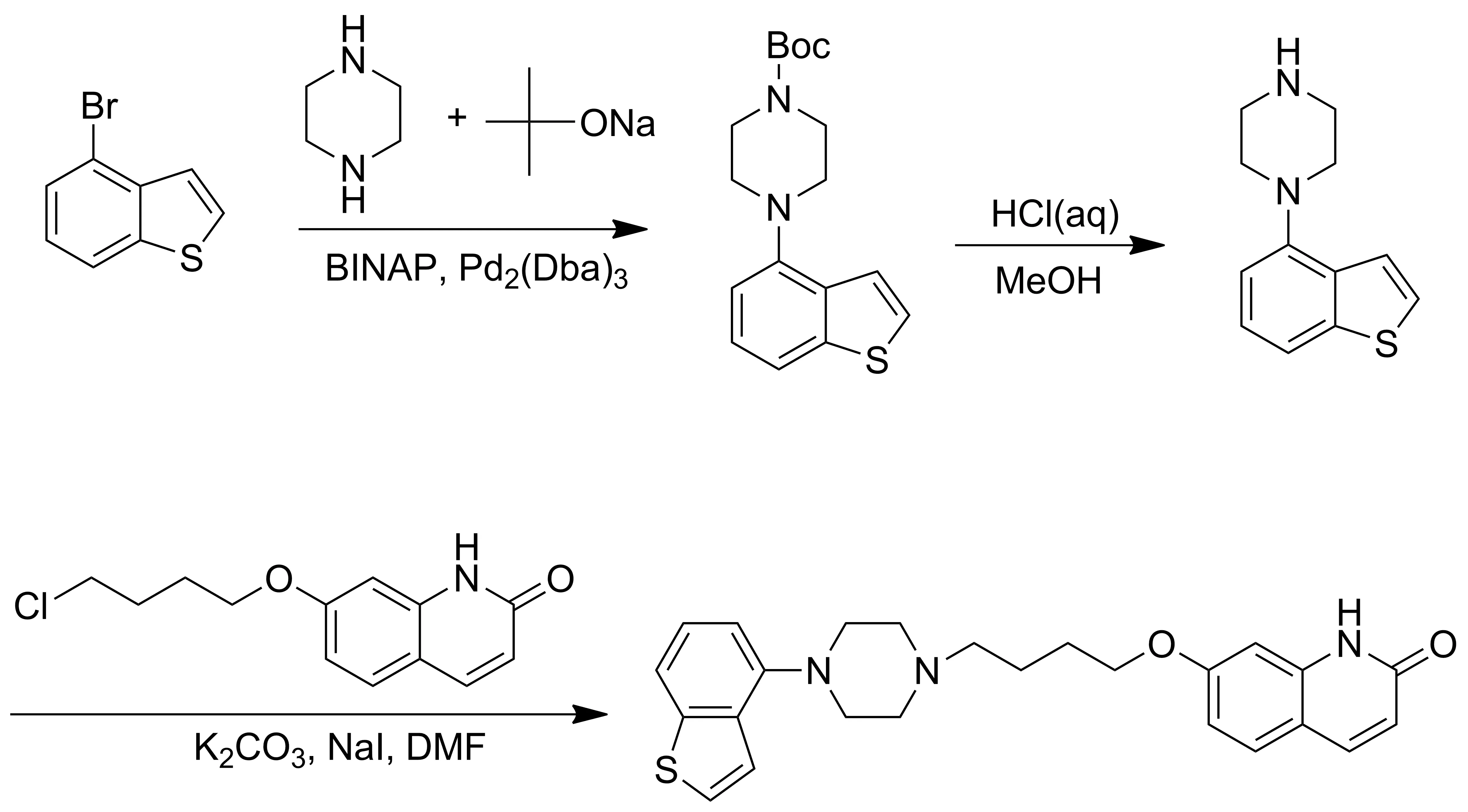
Scheme 4
Vellanki等[6]对Jagtap等的合成路线进行了改进(Scheme 2),但该方法还是存在不足。如原料价格昂贵、生产工序较为繁琐、生产周期长及所用试剂毒性大等问题,特别是使用了甲磺酰氯,对设备腐蚀大,环境不友好且不适合于百克级别的工业化生产。因此,该方法仍有改进的空间。
Rajan等[7]、郑永勇等[8]及孙明哲等[9]采取类似的方法(Scheme 3)合成依匹哌唑。以7-羟基喹啉-2-(1H)酮为起始原料,先和溴氯丁烷反应得4-氯丁氧喹啉酮,再和哌嗪盐酸盐或Boc-哌嗪反应得7-(4-(哌嗪-1-基)丁氧)喹啉-2-(1H)酮后再与4-溴苯并噻吩反应得到依匹哌唑。该法的缺点是接哌嗪一步收率低、副产物较多、后处理繁琐、需要过量的哌嗪化合物,不便于工业化生产。
大冢制药有限公司[10]合成依匹哌唑的原始路线为:通过7-(4-氯丁氧基)-2-(1H)-喹啉酮中间体或其类似物,与4-(1-哌嗪基)苯并[b]噻吩中间体偶联得依匹哌唑(Scheme 4)。该法比较普遍且适合工业化生产,但在合成4-(1-哌嗪基)苯并[b]噻吩中间体时存在哌嗪氮上氢分别被苯并噻吩取代的可能,且在合成7-(4-氯丁氧基)-2-(1H)-喹啉酮中间体时,所用的1-溴-4-氯丁烷选择性较差。
徐奎等[11]以7-羟基-3,4-二氢-2-(1H)-喹啉酮为起始原料,与4-溴丁醇反应得到7-(4-羟基丁氧基)-3,4-二氢-2-(1H)-喹啉酮中间体,再和甲磺酰氯反应生成带有易于离去基团的中间体,与4-(1-哌嗪基)苯并[b]噻吩中间体反应后采用2,3-二氯-5,6-二氰基对苯醌(DDQ)脱氢得到依匹哌唑。该方法中使用的甲磺酰氯对设备腐蚀性强,DDQ毒性大,后处理困难,对环境不友好,限制了该方法的工业化生产。
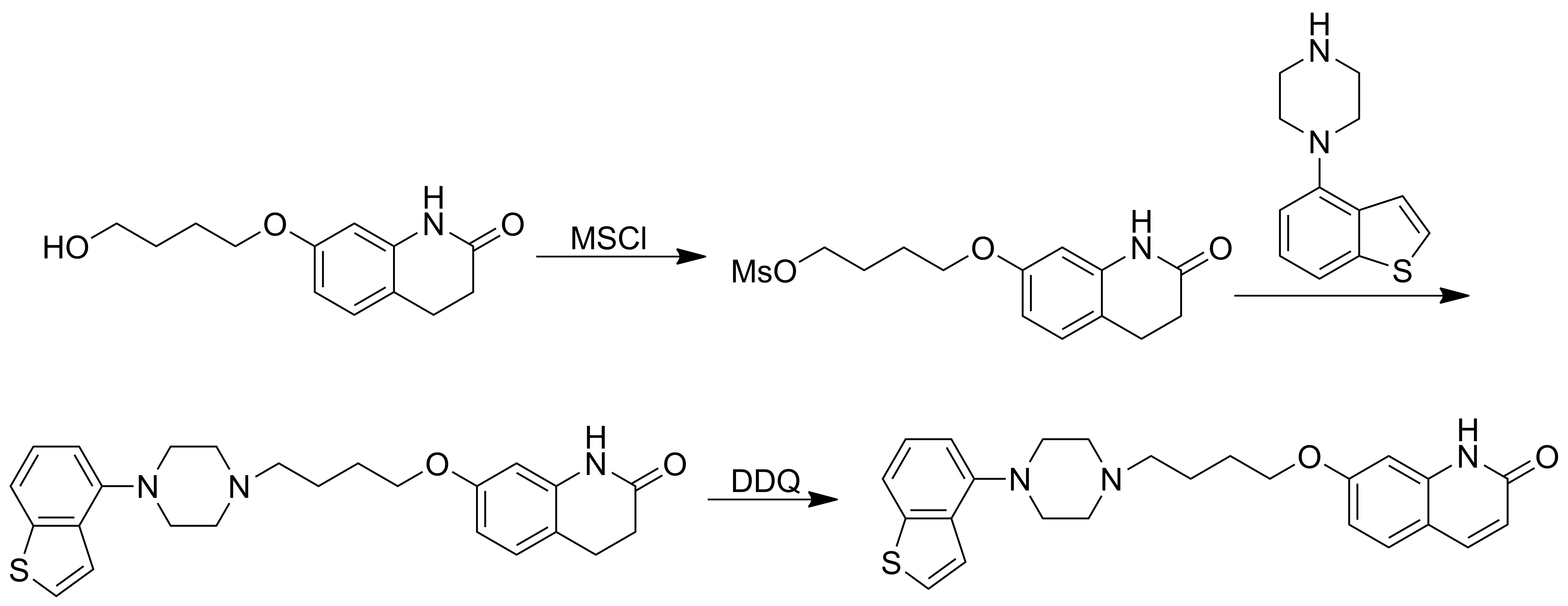
Scheme 5
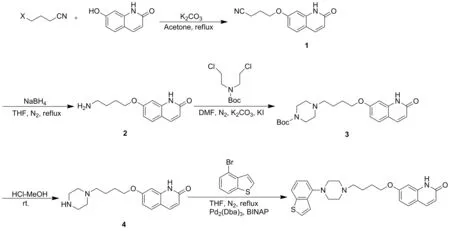
Scheme 6
本文在文献的基础上,设计了依匹哌唑新的合成工艺路线,该路线原料易得、易于操作、环境友好、便于工业化,其收率和纯度较高。将4-取代丁腈和7-羟基2-(1H)-喹啉酮在丙酮中回流反应得到化合物1;产物精制后加入还原剂或催化剂,N2保护下于含碘溶剂中回流反应将化合物1氰基还原胺化得到化合物2;产物精制后和Boc-双氯乙基亚胺在碱性条件下回流反应得到化合物3,反应溶剂可以是二甲基亚砜、乙腈、N,N-二甲基甲酰胺、N-甲基吡咯烷酮、乙二醇单甲醚或正丁醇;化合物3在室温下于酸性溶液中脱保护后得化合物4;产物精制后加入催化剂和配体,和4-溴苯并噻吩在N2保护下回流反应得到依匹哌唑(Scheme 6)。整个工艺路线不涉及高温、高压、低温等特殊反应条件,其中第二步的产物后处理较简单,只需氰基还原胺化即可用于下一步反应,第三步没有使用Boc保护哌嗪参与反应,便于工业化生产;第四步脱保护基采用氯化氢的醇溶液,降低产物的溶解度,可有效提高产物收率。
1 实验部分
1.1 仪器与试剂
JNM-ECZ400S/L1型核磁共振波谱仪(DMSO为溶剂,TMS为内标);Q Exactive Focus型质谱仪;ZF-1型三用紫外分析仪。
7-羟基2-(1H)-喹啉酮,4-溴丁腈,无水碳酸钾,硼氢化钠(分析纯,上海达瑞精细化学品有限公司);Boc-双氯乙基亚胺,碘化钾(分析纯,阿拉丁化学试剂);4-溴苯并噻吩,叔丁醇钾,1,1′-联萘-2,2′-双二苯膦,钯催化剂(分析纯,上海达瑞精细化学品有限公司);其余所用试剂均为分析纯。
1.2 合成
(1) 化合物1的合成
将0.0130 mol(1.924 g)4-溴丁腈加入到50 mL反应瓶中,依次加入0.0120 mol(1.930 g)7-羟基2-(1H)-喹啉酮、0.0060 mol(0.829 g)无水碳酸钾和20 mL丙酮,搅拌下升温至回流,保温反应至终点(TLC跟踪反应,展开剂:二氯甲烷 ∶甲醇=10 ∶1,V∶V)。冷却至室温,抽滤,滤饼用丙酮(2×10 mL)洗涤,合并滤液和洗液,减压蒸除溶剂,剩余物加入10 mL石油醚,充分搅拌,置于冰箱过夜。抽滤,滤饼用石油醚(2×5 mL)洗涤,恒温干燥后得白色固体化合物1,收率78.12%,纯度98.16%;1H NMR(DMSO-d6, 400 MHz)δ: 11.57(s, 1H, NH), 7.77(d,J=7.8 Hz, 1H, PhH), 7.53(d,J=7.5 Hz, 1H, PhH), 6.76~6.75(m, 2H, PhH), 6.27(d,J=6.3 Hz, 1H, PhH), 4.02(t,J=4.0 Hz, 2H, CH2), 2.65(t,J=2.6 Hz, 2H, CH2), 2.03~2.00(m, 2H, CH2); MS(ESI)m/z: calcd for C13H12N2O2{[M+H]+}228.25, found 228.46。
(2) 化合物2的合成
将1.140 g(5.0000 mmol)化合物1加入到100 mL三口反应瓶中,加入30 mL四氢呋喃,搅拌下冷却至0 ℃,加入1.000 g(27.0000 mmol)硼氢化钠,N2保护下,于0 ℃滴加3.000 g(12.0000 mmol)碘的20 mL四氢呋喃溶液,2~3 h滴毕。升温至回流,保温反应至终点(TLC跟踪反应,展开剂:二氯甲烷 ∶甲醇=5 ∶1,V∶V)。冷却至0 ℃,滴加3 N盐酸淬灭,直到无气体放出为止,用3 N的氢氧化钠溶液中和,产生沉淀,抽滤,滤饼用无水乙醇(2×10 mL)洗涤,恒温干燥后得黄色固体,收率96.55%,纯度98.25%;1H NMR(DMSO-d6, 400 MHz)δ: 11.82(s, 1H, NH), 7.76(d,J=7.7 Hz, 1H, PhH), 7.50(d,J=7.5 Hz, 1H, PhH), 6.75~6.71(m, 2H, PhH), 6.26(d,J=6.2 Hz, 1H, PhH), 4.51(s, 1H, NH2), 3.95(t,J=4.0 Hz, 2H, CH2), 2.58~2.54(m, 2H, CH2), 1.71~1.69(m, 2H, CH2), 1.49~1.46(m, 2H, CH2);13C NMR(DMSO-d6, 100 MHz)δ: 162.9, 161.3, 141.3, 140.7, 130.2, 119.0, 114.1, 111.4, 99.4, 68.0, 41.2, 29.1, 26.5; MS(ESI)m/z: calcd for C13H16N2O2{[M+H]+}232.28, found 232.73。
(3) 化合物3的合成
将0.232 g(1.0000 mmol)化合物2加入到反应瓶中,再加入0.266 g(1.0000 mmol)Boc-双氯乙基亚胺和N,N-二甲基甲酰胺5 mL,搅拌下加入0.332 g(2.0000 mmol)碘化钾和0.415 g(3.0000 mmol)无水碳酸钾。N2保护下升温至70 ℃,保温反应至终点(TLC跟踪反应,展开剂:氨水 ∶二氯甲烷 ∶甲醇=1∶6 ∶18,V∶V)。冷却至室温,搅拌下倒入15 mL冰水中,抽滤,滤饼水洗,恒温干燥得类白色固体,收率80.90%,纯度97.89%;1H NMR(DMSO-d6, 400 MHz)δ: 12.51(s, 1H, NH), 7.75(d,J=7.8 Hz, 1H, PhH), 7.45(d,J=7.6 Hz, 1H, PhH), 6.83~6.79(m, 2H, PhH), 6.56(d,J=6.6 Hz, 1H, PhH), 4.10(t,J=4.1 Hz, 2H, CH2), 3.45~3.43(m, 2H, CH2), 2.44~2.41(m, 2H, CH2), 1.72~1.69(m, 2H, CH2), 1.48(s, 9H, 3CH3); MS(ESI)m/z: calcd for C22H31N3O4{[M+H]+}401.51, found 401.64。
(4) 化合物4的合成
将0.402 g(1.0000 mmol)化合物3加入到反应瓶中,加入饱和氯化氢的甲醇10 mL,于室温搅拌2 h,置于冰箱冷冻,抽滤,甲醇洗涤滤饼,恒温干燥得白色固体,收率73.10%,纯度98.52%;1H NMR(DMSO-d6, 400 MHz)δ: 11.59(s, 1H, NH), 7.76(d,J=7.8 Hz, 1H, PhH), 7.49(d,J=7.5 Hz, 1H, PhH), 6.75~6.72(m, 2H, PhH), 6.26(d,J=6.2 Hz, 1H, PhH), 4.20(t,J=4.2 Hz, 2H, CH2), 3.98(t,J=4.0 Hz, 2H, CH2), 3.53(t,J=3.5 Hz, 2H, CH2), 3.19(t,J=3.2 Hz, 2H, CH2), 2.68(t,J=2.7 Hz, 2H, CH2), 2.55(t,J=2.6 Hz, 2H, CH2), 1.84(s, 1H, NH), 1.72~1.69(m, 2H, CH2), 1.56~1.51(m, 2H, CH2); MS(ESI)m/z: calcd for C17H23N3O2{[M+H]+}301.39, found 301.91。
(5) 依匹哌唑的合成
将0.0135 mol(2.880 g)4-溴苯并噻吩加入到100 mL反应瓶中,分别加入0.0135 mol(4.070 g)化合物4、叔丁醇钾0.0190 mol(2.120 g)、 0.130 g 1,1′2,2′双二苯膦、0.120 g钯催化剂和50 mL四氢呋喃,N2保护下搅拌升温至回流,保温反应至终点(TLC跟踪反应,展开剂:二氯甲烷 ∶甲醇=20 ∶1,V∶V)。冷却至室温,减压蒸除溶剂,室温下加入30 mL水和30 mL二氯甲烷,搅拌下滴加2 N的盐酸调节pH值至酸性,继续搅拌40 min,过滤,滤液分层,有机相用浓盐酸酸化,加入甲醇,充分搅拌,抽滤,二氯甲烷洗涤滤饼,真空干燥得类白色固体,粗产物的收率可达92.56%,纯度92.78%,经75.00%乙醇重结晶后目标产物收率为68.49%,纯度99.30%,单杂含量小于0.10%;1H NMR(DMSO-d6, 400 MHz)δ: 11.54(s, 1H, NH), 7.60(d,J=7.6 Hz, 1H, PhH), 7.64(d,J=7.6 Hz, 1H, PhH), 7.55(d,J=7.6 Hz, 1H, PhH), 7.51(d,J=7.5 Hz, 1H, PhH), 7.35(d,J=7.3 Hz, 1H, PhH), 7.23(t,J=7.2 Hz, 1H, PhH), 6.82(d,J=6.8 Hz, 1H, PhH), 6.76~6.74(m, 2H, PhH), 6.26(d,J=6.3 Hz, 1H, PhH), 4.00(t,J=4.2 Hz, 2H, CH2), 3.02~2.98(m, 4H, CH2), 2.57~2.54(m, 4H, CH2), 2.37(t,J=2.6 Hz, 2H, CH2), 1.74~1.71(m, 2H, CH2), 1.59~1.57(m, 2H, CH2); MS(ESI)m/z: calcd for C25H27N3O2S{[M+H]+}433.57, found 433.73。
2 结果与讨论
2.1 醚化物1的合成
在化合物1的合成过程中,考察了缚酸剂和取代丁腈对醚化物收率的影响,结果如表1所示。将4-溴丁腈换成4-羟基丁腈,其它反应步骤、原料和反应的量不改变,粗产物的收率为75.36%,精制后纯度95.72%;原料的改变和醚化物收率的改变可能与离去基团的离去能力有关。将无水碳酸钾换为四氢吡咯,其它反应步骤、原料和反应的量不改变,粗产物的收率为63.25%,精制后纯度97.62%;从实验结果来看,无水碳酸钾作为缚酸剂,虽然是非均相反应,但效果比四氢吡咯更好,因此在实际合成过程中采用了无水碳酸钾作缚酸剂。
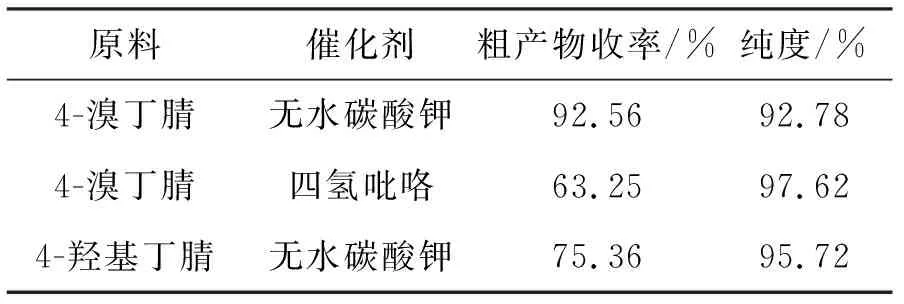
表1 原料和催化剂对化合物1合成的影响
2.2 氨化物2的合成
在氨化物2的合成过程中,对醚化物1的氰基还原方法进行考察,采用金属氢化物和催化氢化法将氰基还原成氨基,结果如表2所示。从表2可以看出,采用硼氢化钠还原氰基的收率更高。
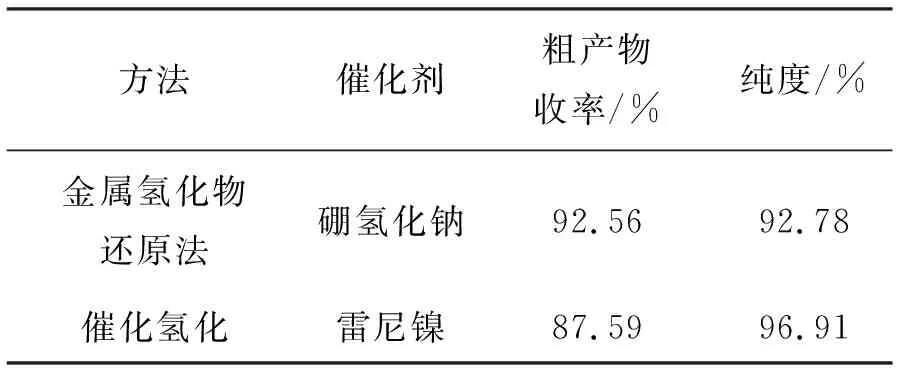
表2 还原方法对化合物2合成的影响
2.3 依匹哌唑的合成
在依匹哌唑的合成过程中,考察了催化剂对目标产物收率的影响,将双亚苄基丙酮钯分别换成钯碳及醋酸钯作催化剂,结果如表3所示。从表3可以看出,采用双亚苄基丙酮作催化剂的收率最佳。
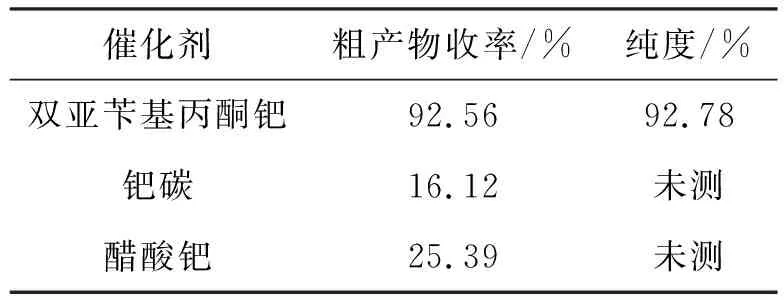
表3 不同催化剂对依匹哌唑收率的影响
本文经过实验条件筛选与重复验证,优选4-卤代丁腈为起始原料,采用碳酸钾作为第1步和第3步反应催化剂,优选金属氢化物还原法将氰基还原为氨基,优选双亚苄基丙酮钯作为目标产物合成的催化剂,该工艺采用普通原料,反应条件温和易控,粗产物的收率可达92.56%,纯度为92.78%,目标产物精制后的收率为68.49%,纯度为99.30%,所用溶剂均可回收,有效降低成本,减少了环境污染。
