基于SLAF-seq技术的舒玛栎群体遗传多样性与遗传结构分析
2022-11-30何旭东郑纪伟教忠意窦全琴黄利斌
何旭东,郑纪伟,教忠意,窦全琴,黄利斌
(江苏省林业科学研究院,江苏 南京 211153)
栎树是壳斗科(Fagaceae)栎属(Quercus)植物的统称。栎树是壳斗科中分化最晚、进化程度最高的类群,全世界有450余种,主要分布于北纬16°~62°地区,在美国、俄罗斯和中国等地区资源最为丰富[1]。我国约有栎树51种,除新疆仅有栽培种外,其余各省均有自然分布[2]。栎树以落叶或常绿乔木为主,通常认为华北地区为落叶栎的分布中心,云贵高原为常绿栎的分布中心[3]。栎树是亚热带至温带阔叶森林重要的建群种和优势种,在森林生态系统中起着不可替代的作用。栎树木材坚硬,纹理美观,为珍贵用材树种;其树形高大,叶色多变,也是重要的园林观赏树种;同时其树皮、树叶、果实等还可作为不同用途的工业原料[4-5]。
尽管我国栎树种质资源丰富,但大多处于野生状态,以天然次生林为主,并与其他树种相互混杂,在种质资源挖掘与品种选育方面几乎为空白,导致乡土栎树在优质用材及园林绿化应用方面较国外差距较大[6-7]。自20世纪90年代末,中国林业科学研究院与江苏省林业科学研究院等科研单位陆续开展系统性的国外栎树引种研究,引进了纳塔栎(Q.nuttallii)、舒玛栎(Q.shumardii)、柳叶栎(Q.phellos)、北方红栎(Q.rubra)、弗吉尼亚栎(Q.virginiana)等树种,对其生长性、适应性、抗逆性和观赏性等进行了评价[8-12],并对无性繁殖技术进行了研究[13]。但总体而言,国内对引种栎树的研究仍局限于常规育种方面,对国外不同栎树种质资源的遗传基础及亲缘关系方面的研究依然比较缺乏。
遗传多样性是物种进化和适应的基础,是抵抗环境变化的重要保障。遗传多样性水平的高低体现物种对环境变化的适应能力,是物种长期存活的决定因素之一。此外,遗传多样性也是遗传结构研究的核心内容,而遗传结构的研究对于种质资源的挖掘、利用和保护具有极其重要的理论与实践意义[14-16]。国内外利用分子标记技术对多个栎树种[17-25]进行了遗传多样性与遗传结构等研究。此外,国外还对分布于美洲大陆的多种栎树进行了群体多样化的研究[26-27]。本研究选取江苏省林业科学研究院前期引进的6个舒玛栎种源,采用SLAF-seq技术开发一批SNP标记,并对不同种源舒玛栎群体进行遗传多样性与遗传结构分析,以期为舒玛栎资源引进、种质保存以及品种选育提供理论依据,也为其他珍贵彩色栎树树种的开发利用提供有益参考。
1 材料与方法
1.1 试验材料和DNA提取
供试材料为江苏省林业科学研究院前期从美国引进的6个舒玛栎种源,每个种源包含5个单株,共30个个体(表1)。此外,还选取5个纳塔栎个体作为外类群一并进行分析。所有个体均定植于江苏省林业科学研究院种质资源圃内。
采集所有个体幼嫩叶片,利用试剂盒(天根生化生物工程有限公司,DP305)提取DNA。用质量分数0.8%琼脂糖凝胶电泳检测DNA的质量,Nanodrop检测DNA的浓度,-20 ℃保存备用。

表1 试验材料Table 1 Experimental materials
1.2 文库构建与SLAF测序
利用北京百迈客生物科技有限公司自主研发的特异性位点扩增片断测序(specific-locus amplified fragment sequencing,SLAF-seq)技术对35个个体进行测序[28]。采用无参考基因组方案,确定限制性内切酶为RsaI,将酶切片段长度在314~414 bp的序列定义为SLAF标签。对SLAF标签进行3′端加A处理,连接Dual-index测序接头建库测序。为评估建库实验的准确性,选用水稻品种‘日本晴’(Oryzasativa‘Nipponbare’,基因组大小为374.30 Mbp,http://rapdb.dna.affrc.go.jp/)作为对照,测序平台为Illumina HiSeq 2500。对测序得到的原始数据进行识别、过滤、质检、评估等分析,获取各个个体的序列(reads)。
1.3 SLAF标签与SNP标记开发
将各个体的reads根据相似性进行聚类,聚类到一起的reads来源于一个SLAF标签。因同一个SLAF标签在不同个体间序列相似度远高于不同SLAF标签间的相似度,因此将在不同个体间有序列差异的SLAF标签定位为多态性标签。以每个SLAF标签中测序深度最高的序列类型作为参考序列,利用bwa将测序reads比对到参考序列上[29],使用GATK和samtools两种方法开发SNP[30-31],将两种方法得到的SNP标记交集作为最终可靠的SNP标记集合,并以完整度>0.8、最小等位基因频率(MAF)>0.05为标准进行过滤。
1.4 数据分析
利用GenAlex 6.5[32]软件计算各个种源有效等位基因数(Ne)、多态位点比例(PPL)、观测杂合度(Ho)、期望杂合度(He)、多态信息含量(PIC)、香农指数(I)、基因多样性指数(H)等遗传多样性参数;利用Arlequin 3[33]软件计算群体内近交系数(FIS)、群体间遗传分化系数(FST)以估计群体遗传分化程度,并进行分子方差分析(analysis of molecular variance,AMOVA)以检测群体内和群体间遗传变异情况;利用MEGA X(https://www.megasoftware.net)软件基于Neighbor-joining算法(Kimura 2模型,1 000次bootstrap)构建群体进化树;利用Admixture[34]软件分析群体结构,设定群体分群数(K值)为1~10进行聚类,根据ΔK峰值的位置来确定分群数;利用Cluster X软件进行主成分分析并聚类。
2 结果与分析
2.1 SLAF标签与SNP标记开发结果
利用SLAF测序技术对35个栎树个体进行高通量测序,共获得50.4 Mb酶切reads,按不同群体统计相关指标。如不同种源栎树SLAF标签与SNP标记统计(表2)所示。

表2 不同种源栎树SLAF标签与SNP标记统计Table 2 Statistics of SLAF tags and SNP markers for different provenances of Quercus spp.
7个群体Q30值变化范围为90.24%~95.12%,最高为PA群体,最低为NT群体,平均为93%,表明测序质量较好。GC含量最高为OH群体(39.35%),最低为PA群体(38.49%),平均为38.9%。酶切共获得4 256 436个多态性的SLAF标签,最多的TX群体平均每个个体134 816.4个,最少的NT群体平均每个个体107 005.2个。每个群体的测序深度从10.62至12.38不等,平均为11。舒玛栎各个群体中个体平均SNP数量相差不大,LA群体中每个个体SNP数量最多,平均为259 700.2个,TX群体最少,平均为236 358个;纳塔栎群体中每个个体SNP数量平均为200 735个。各群体间SNP完整度变化范围为65.85%~85.20%,平均为79.29%,SNP杂合率变化范围为8.67%~16.01%,平均为14.15%,均为LA群体最大,NT群体最小。
2.2 舒玛栋群体遗传多样性与遗传分化
舒玛栎群体遗传多样性分析结果(表3)显示:6个种源平均有效等位基因数为1.31个;多态性位点比例平均为49.21%,最高为PA种源(57.44%),最低为OH种源(43.34%);各种源间观测杂合度与期望杂合度变化范围不大,分别为0.13~0.16和0.17~0.21;PIC值变化范围为0.13~0.17,平均为0.15;香农指数PA种源最高,为0.39,OH种源最低,为0.30,平均为0.34;各种源间Nei’s基因多样性指数变化范围较小,平均为0.09;群体内的近交系数最大为MO与MS种源,为0.26,最低为LA与OH种源,为0.10,种源间平均为0.19。

表3 不同种源舒玛栎群体遗传多样性Table 3 Genetic diversity of the six different provenances of Quercus shumardii
舒玛栎不同种源间的Nei’s遗传距离变化范围为0.08~0.18,其中MS种源与TX种源遗传距离最远,为0.18,LA种源与MO种源以及MO种源与PA种源遗传距离最近,为0.08(表4)。不同种源群体间遗传分化系数FST变化范围为0.15~0.39,其中TX种源与MS种源以及TX种源与OH种源群体间遗传分化系数最高,为0.39;其次为OH种源与MS种源以及TX种源与LA种源,为0.35;PA种源与MO种源群体间遗传分化系数最低,为0.15。由分子方差分析(AMOVA)可知,舒玛栎遗传变异主要来自群体内(84.88%),群体间的遗传变异占总变异的15.12%(表5)。

表4 不同种源舒玛栎群体间Nei’s遗传距离与遗传分化系数Table 4 Pairwise differentiation (FST) and genetic distance among provenances of Quercus shumardii

表5 不同种源舒玛栎群体分子方差分析Table 5 Analyses of molecular variances of the six different provenances of Quercus shumardii
2.3 舒玛栎不同种源的遗传结构与亲缘关系
利用Admixture软件分析舒玛栎群体遗传结构,分别假设舒玛栎30个个体的分群数(K值)为1~10进行聚类,根据交叉验证错误率的谷值确定最优分群数为3(图1A),表明30个个体来自3个类群。如图1B所示,本研究的30个个体可以划分为3个群,类群1(红色基因池)主要包括TX种源,类群2(绿色基因池)主要包括LA、MO、OH和PA种源,类群3(黄色基因池)主要包括MS种源。
为进一步探讨舒玛栎不同种源个体间的亲缘关系,以5个纳塔栎个体作为外类群,使用群体SNP标记计算个体间遗传距离,并利用MAGA软件基于neighbor-joining算法构建30个舒玛栎个体的进化树,同时利用Cluster X软件进行主成分分析(图2)。如图2A所示,35个栎树个体聚成两大类,其中外类群纳塔栎5个个体单独聚成一类,30个舒玛栎个体聚成一大类,与传统的分类学一致。6个种源的舒玛栎群体也基本各自单独聚成一小类。图2B所示的三维主成分聚类图也可以辅助亲缘关系分析,通过空间距离的远近判断不同个体间的亲缘关系。
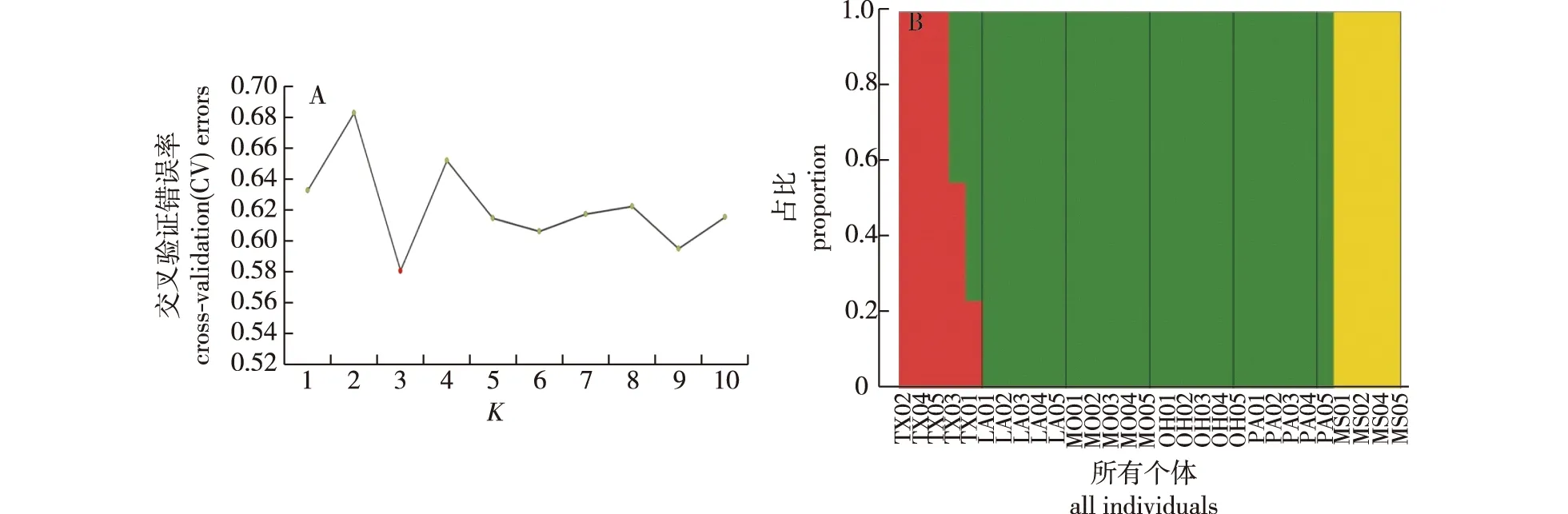
图1 舒玛栎群体最佳类群数与遗传结构Fig.1 The rational groups number and genetic structure of Quercus shumardii

图2 35个栎树个体进化树(A)与主成分分析(B)Fig.2 Phylogenetic tree and principal components analyses of 35 individuals of Quercus
3 讨 论
利用SLAF-seq技术对美国引进的6个种源舒玛栎群体进行遗传多样性和遗传结构分析,旨在开发一批可用的SNP标记,为舒玛栎种质资源的开发、利用与保护提供理论依据,也为其他珍贵彩色树种的研究提供有益参考。因受前期引种条件限制,本研究中舒玛栎群体每个种源只有5个个体,样本量较小,部分研究结果可能不能完全反映不同种源间大空间尺度的群体遗传多样性与遗传结构的真实情况,但有研究表明,较小的群体仍可有效地估算遗传多样性等相关参数[35]。如在Q.susber遗传多样性与遗传结构研究中,6个群体的样本量均为5个个体,3个群体的样本量为8~10个[17];在细叶桉(Eucalyptustereticornis)9个群体遗传多样性分析中,其中1个群体只有4个样本,3个群体包含5个样本,1个群体有7个样本[35];在木荷(Schimasuperba)遗传多样性分析中,多个群体的样本量也都在10个以下[36]。总体而言,群体中个体数量多少为宜没有明确的定论,Singh等[37]认为有的群体5~6个个体即可满足遗传多样性分析的需要,有的群体则需要12个以上的个体,而且样本量的大小也与物种本身的生物学特性以及取样策略有关。
多态性位点比例是反映遗传多态性的重要指标。本研究中舒玛栎群体的平均多态性SNP位点比例为49.21%,高于ISSR标记检测的Q.susber(36.99%)[17]与蒙古栎(Q.mongolica)(45.24%)[38]群体,略低于ISSR标记检测的Q.libani(52.61%)群体[21],而在Q.infectoria群体中,基因组SSR标记平均多态性位点比例高达100%,叶绿体SSR标记却只有19.19%,可能是由于叶绿体SSR过于保守,标记本身多态性较低造成。期望杂合度He常用来衡量群体遗传多样性的高低,He越高,群体一致性就越低,遗传多样性就越丰富。本研究中,舒玛栎群体平均期望杂合度He为0.19,远低于SSR标记检测的Q.infectoria(平均He为0.75)[19]、Q.rubra和Q.ellipsoidalis(平均He为0.78)[20]以及栓皮栎(Q.variabilis)(平均He为0.707)[22]群体。推测一方面可能与样本大小有关,另一方面可能与标记类型有关,如同样采用SNP标记检测的油棕群体(平均He为0.29~0.32)[39]和榔榆(Ulmusparvifolia)群体(平均He为0.33)[40],其He值与本研究较为接近。类似的,又如多态信息含量PIC值,SSR标记[22]也明显高于SNP标记[39-40]。此外,本研究中舒玛栎群体的香农指数I平均为0.34,高于ISSR标记检测的Q.susber(I= 0.168)[17]与Q.libani群体(I= 0.25)[21],而基因组SSR标记检测的Q.infectoria群体I值高达1.55,叶绿体SSR标记I值却只有0.09。前期的研究结果也表明:当某一标记的等位基因变异数较多时,检测的有效等位基因、杂合度、香农指数等参数也较高[14]。由此可见,标记类型与效率对遗传多样性参数估算的影响较大,一般应选择多态性较高的标记才能最大程度地揭示群体遗传多样性的真实水平。
F统计量可以反映群体遗传结构的变化,主要受突变、交配、漂变及选择等因素的影响,一般用群体内近交系数FIS、群体总近交系数FIT和遗传分化系数FST来衡量。本研究中,舒玛栎各种源平均近交系数FIS为0.19,明显高于Q.semiserrata(平均FIS为0.01)[18]与栓皮栎(平均FIS为0.044)[22]群体,推测可能是由于样本量较小、个体间亲缘关系较近造成的。本研究中,不同种源群体间遗传分化系数FST变化范围为0.15~0.39,也高于Q.semiserrata(平均FST为0.12)[18]、Q.ellipsoidalis(平均FST为0.04)[20]以及栓皮栎(平均FST为0.063)[22]群体。通常认为FST值为0.15~0.25,群体间存在较大遗传分化;FST值在0.25以上,群体间有很大的遗传分化[41]。由方差AMOVA分析也可以看出,舒玛栎群体间变异贡献率达到15.12%,表明群体间存在较大的遗传分化。此外,舒玛栎群体的遗传变异主要来源于个体间,因此在舒玛栎品种选育工作中,应侧重于群体内单个优树的选择。
