Single-nucleus transcriptomic profiling of multiple organs in a rhesus macaque model of SARS-CoV-2 infection
2022-11-29QiangMaWenjiMaTianZhangSongZhaoboWuZeyuanLiuZhenxiangHuJianBaoHanLingXu3BoZengBosongWangYinuoSunDanDanYu3QianWuYongGangYao3YongTangZheng3XiaoqunWang
Qiang Ma, Wenji Ma, Tian-Zhang Song, Zhaobo Wu, Zeyuan Liu, Zhenxiang Hu, Jian-Bao Han, Ling Xu3,,5,Bo Zeng, Bosong Wang, Yinuo Sun, Dan-Dan Yu3,,5, Qian Wu,8, Yong-Gang Yao3,,5,*, Yong-Tang Zheng3,,5,*,Xiaoqun Wang,,8,9,*
1 State Key Laboratory of Brain and Cognitive Science, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China
2 University of Chinese Academy of Sciences, Beijing 100049, China
3 Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences, KIZ-CUHK Joint Laboratory of Bioresources and Molecular Research in Common Diseases, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming,Yunnan 650223, China
4 Kunming National High-Level Biosafety Research Center for Non-Human Primates, Center for Biosafety Mega-Science, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650107, China
5 National Resource Center for Non-Human Primates, National Research Facility for Phenotypic & Genetic Analysis of Model Animals(Primate Facility), Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650107, China
6 LivzonBio, Inc., Zhuhai, Guangdong 519045, China
7 State Key Laboratory of Cognitive Neuroscience and Learning, Beijing Normal University, Beijing 100875, China
8 IDG/McGovern Institute for Brain Research, Beijing Normal University, Beijing 100875, China
9 Advanced Innovation Center for Human Brain Protection, Beijing Institute for Brain Disorders, Capital Medical University, Beijing 100069,China
ABSTRACT Infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes diverse clinical manifestations and tissue injuries in multiple organs.However, cellular and molecular understanding of SARS-CoV-2 infection-associated pathology and immune defense features in different organs remains incomplete. Here, we profiled approximately 77 000 single-nucleus transcriptomes of the lung, liver,kidney, and cerebral cortex in rhesus macaques(Macaca mulatta) infected with SARS-CoV-2 and healthy controls. Integrated analysis of the multiorgan dataset suggested that the liver harbored the strongest global transcriptional alterations. We observed prominent impairment in lung epithelial cells, especially in AT2 and ciliated cells, and evident signs of fibrosis in fibroblasts. These lung injury characteristics are similar to those reported in patients with coronavirus disease 2019 (COVID-19).Furthermore, we found suppressed MHC class I/II molecular activity in the lung, inflammatory response in the liver, and activation of the kynurenine pathway,which induced the development of an immunosuppressive microenvironment. Analysis of the kidney dataset highlighted tropism of tubule cells to SARS-CoV-2, and we found membranous nephropathy (an autoimmune disease) caused by podocyte dysregulation. In addition, we identified the pathological states of astrocytes and oligodendrocytes in the cerebral cortex, providing molecular insights into COVID-19-related neurological implications. Overall, our multi-organ single-nucleus transcriptomic survey of SARS-CoV-2-infected rhesus macaques broadens our understanding of disease features and antiviral immune defects caused by SARS-CoV-2 infection,which may facilitate the development of therapeutic interventions for COVID-19.
Keywords: SARS-CoV-2; Rhesus macaque;Animal model; Single-nucleus RNA sequencing;Antiviral immune defects; Multiple organs
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARSCoV-2), the causative agent of coronavirus disease 2019(COVID-19), continues to threaten global health, with more than 620 million confirmed cases so far (as of 1 November 2022; https://www.who.int/emergencies/diseases/novelcoronavirus-2019). Patients may present with fever, dry cough, and dyspnea, with some developing acute respiratory distress syndrome (ARDS), potentially leading to death in critical cases (Wang et al., 2020a; Wiersinga et al., 2020).Histopathological examination of autopsy samples shows diffuse alveolar damage (DAD) and exudative inflammation(Liu et al., 2020a). Liver and kidney function abnormalities have also been observed in COVID-19 patients, as indicated by elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, proteinuria, and hematuria(Cheng et al., 2020; Fu et al., 2021). In addition, SARS-CoV-2 infection can affect the central nervous system (CNS),resulting in neurological symptoms in some patients (Mao et al., 2020). Cognitive deficits (Liu et al., 2021) and accelerated aging (Cao et al., 2022), including aging signatures in the brain (Mavrikaki et al., 2021), have also been reported in people with COVID-19.
Recently, postmortem organ tissue specimens have been used to analyze COVID-19 pathology at single-cell resolution(Delorey et al., 2021; Melms et al., 2021; Ren et al., 2021;Wang et al., 2021; Yang et al., 2021). However, human subjects show considerable variation in exposure level,exposure route, sampling time relative to exposure, and background health conditions. Postmortem interval may also have an adverse effect on tissue quality. Furthermore,obtaining age-matched healthy tissue controls is a challenge.The limited accessibility of autopsy specimens from patients who died of COVID-19 and matched healthy controls has restricted in-depth pathological study in human subjects.
These limitations can be counteracted by using animal models, although severe disease observed in humans cannot be completely recapitulated. Animals, including monkeys, tree shrews, and ferrets, are permissive to SARS-CoV-2 infection(Deng et al., 2020; Fan et al., 2022; Kim et al., 2020; Muñoz-Fontela et al., 2020; Munster et al., 2020; Shan et al., 2020;Song et al., 2021; Song et al., 2020; Xu et al., 2020), with various animal models developed to test vaccines (Gao et al.,2020; Wang et al., 2020b) and understand disease progression and immune responses (Lee et al., 2021; Nelson et al., 2022). Rhesus macaques are a commonly used nonhuman primate model for studies on human respiratory viral diseases (Skinner et al., 2014; Yao et al., 2014). Their inoculation with SARS-CoV-2 can result in mild-to-moderate respiratory disease (Deng et al., 2020; Munster et al., 2020;Shan et al., 2020; Song et al., 2020), suggesting that rhesus macaques may be a suitable non-human primate model to mimic COVID-19-associated pathology and expand knowledge about this disease.
Current understanding of SARS-CoV-2 infection-induced cellular and molecular pathology remains incomplete (Merad et al., 2022). To identify cell types critical for the tropism observed in affected organs and to analyze infection-induced dysfunction in a cell-type-specific manner, we profiled singlenucleus transcriptomic atlases of visceral organs (i.e., lung,liver, and kidney) and the CNS (i.e., cerebral cortex) from SARS-CoV-2-infected rhesus macaques. Compared with the healthy macaques, infection-related injuries in the macaque lungs were similar to those reported in human patients,indicating that macaques are reliable models with which to recapitulate SARS-CoV-2-caused pathology. Furthermore, we found several previously unidentified infection-associated disorders and antiviral immune defects, providing insights into potential therapeutic interventions for COVID-19.
MATERIALS AND METHODS
Animals and ethics statement
Chinese rhesus macaques (Macaca mulatta) were sourced from the Laboratory Animal Center, Kunming Institute of Zoology (KIZ), Chinese Academy of Sciences (CAS). All animal experiment procedures were performed at the Kunming National High-Level Biosafety Research Center for Non-Human Primates, KIZ, CAS, and in accordance with the guidelines approved by the Ethics Committee of the Kunming Institute of Zoology (approval number: IACUC20005).
SARS-CoV-2 challenge in rhesus macaques
The SARS-CoV-2 strain 107 (accession number:NMDCN0000HUI) was provided by the Guangdong Provincial Center for Disease Control and Prevention (Guangzhou,China), and has been described in our previous studies (Song et al., 2021; Song et al., 2020). The virus was propagated in African green monkey kidney epithelial cells (Vero-E6) (ATCC,No. 1586) and titrated. The macaques were intranasally (40%)and endotracheally (60%) challenged with 107TCID50(50%tissue culture infectious dose) of SARS-CoV-2 and euthanized on the 7th day post-infection (dpi). All seven lung lobes, liver,kidney, and cerebral cortex (prefrontal) were collected and stored in ultra-low temperature refrigerators or immediately fixed in 4% (wt/vol) formaldehyde solution.
Quantitative real-time polymerase chain reaction (qRTPCR) for viral load detection
Lung tissue samples were homogenized, and RNA was extracted using TRIzol Reagent (Thermo, USA). For detection of SARS-CoV-2 RNA, a THUNDERBIRD Probe One-Step qRT-PCR Kit (TOYOBO, Japan) was used in accordance with the manufacturer’s protocols. Primers and probes included:forward primer 5"-GGGGAACTTCTCCTGCTAGAAT-3",reverse primer 5"-CAGACATTTTGCTCTCAAGCTG-3", and probe FAM-TTGCTGCTGCTTGACAGATT-TAMRA-3". In each run, serial dilutions of the SARS-CoV-2 RNA reference standard (National Institute of Metrology, China) were run in parallel to calculate viral copy numbers in each sample.
Tissue processing and nuclei isolation for lung, kidney,and liver tissues
We dissolved one tablet of cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche, #11873580001, Switzerland) into 1 mL of Nuclei EZ lysis buffer from a Nuclei EZ Prep Isolation Kit (Sigma, Cat #NUC-101, USA), named cOmplete stock(10X). We then prepared lysis buffer containing 1.8 mL of EZ lysis buffer supplemented with 200 μL of cOmplete stock and 50 μL of Ambion™ RNase Inhibitor (Invitrogen, AM2684,USA). We added 2 mL of lysis buffer to C tubes (Miltenyi Biotec, #130-093-237, Germany), which were maintained on ice.
Frozen visceral organ tissue specimens (lower lung, liver,and kidney) were taken out from the ultra-low temperature refrigerator, placed in a clean dish on ice, and thawed for 2 min. We injected 1 mL of lysis buffer into the tissue to “inflate”it with a syringe and moved the needle during injection to distribute the solution evenly. The tissues were then cut into small pieces with scissors. The minced tissue pieces and solution were transferred into a C tube containing 2 mL of the prepared lysis buffer. The C tube was closed and inverted to ensure that all pieces were at the base. The C tube was placed on a gentleMACS™ Dissociator (Miltenyi Biotec, #130-093-235, Germany). The m_lung_01 program was run completely, followed by the m_lung_02 program for no more than 20 s, then stopped. The C tube was immediately placed on ice. The homogenate was foamy and contained some small pieces of tissue that had not been dissociated. The foam and supernatant were discarded using wide bore tips after centrifuging the C Tube in a swinging bucket rotor centrifuge at 500 ×gfor 5 min at 4 °C. The pellet was resuspended in 2-4 mL of resuspension buffer (consisting of 1×phosphatebuffered saline (PBS), 0.1% bovine serum albumin (BSA), and 0.5 U/μL Ambion™ RNase Inhibitor (Invitrogen, AM2684,USA)). The suspension was filtered through a 70 μm cell strainer into a new 15 mL centrifuge tube to remove the remaining tissue pieces. Nuclei were pelleted by centrifugation at 300 ×gfor 3 min at 4 °C to avoid blebbing and clumping of nuclei. The supernatant was aspirated thoroughly, and the isolated nuclei were resuspended in 1-2 mL of resuspension buffer. The nuclear suspension was then filtered through a 35 μm cell strainer for further cleaning. The nuclei were counted with a hemocytometer, and nuclear concentration was adjusted to 700-1 200/μL.
Tissue processing and nuclei isolation for cerebral cortex tissues
Frozen prefrontal cortex tissue blocks were taken out from the ultra-low temperature refrigerator and minced into small pieces in a clean dish on ice. The tissue pieces were transferred to pre-chilled glass dounce tissue grinders (Sigma,Cat #D8938, USA) for homogenization in 2 mL of cold Nuclei EZ lysis buffer (Sigma, Cat #NUC-101, USA) on ice, 20 times with pestle A followed by 20 times with pestle B. The homogenate was transferred into a 15 mL centrifuge tube with an additional 2 mL of cold Nuclei EZ lysis buffer, then incubated on ice for 5 min before centrifugation (500 ×gfor 5 min at 4 °C). The supernatant was discarded, and the sediment was resuspended in 4 mL of cold Nuclei EZ lysis buffer, then incubated on ice. After centrifugation (500 ×gfor 5 min at 4 °C) and removal of the supernatant, the primitive nuclear products were cleaned via density gradient centrifugation to remove debris. The sediment was resuspended in 3 100 μL of cold PBS and filtered through a 70 μm cell strainer into a new tube. Cold debris removal solution(900 μL) (Miltenyi Biotec, #130-109-398, Germany) was added to the suspension and mixed well. The mixture was overlaid with 4 mL of cold PBS gently, followed by centrifugation at 3 000 ×gfor 10 min at 4 °C with full acceleration and full brake. The debris aggregated together to form a cloud-like layer at the interphase during centrifugation.The two top phases were discarded, followed by the addition of cold PBS and gentle inversion of the tube three times. The suspension was pelleted by centrifugation at 1 000 ×gfor 5 min at 4 °C. The supernatant was aspirated completely. The nuclear sediment was twice washed with 2 mL of nuclei suspension buffer (NSB, consisting of 1× PBS, 0.1% BSA,and 0.5 U/μL Ambion™ RNase Inhibitor (Invitrogen, AM2684,USA)) for further purification. Finally, the isolated nuclei were resuspended in an appropriate volume of NSB. The nuclei were counted with a hemocytometer, with the concentration adjusted to 700-1 200/μL.
Single-nucleus RNA-seq
The nuclear suspensions were loaded onto Chromium Single-Cell Chip B (10X Genomics, USA) to generate single-cell Gel Beads-in-Emulsion (GEMs) using the Chromium Single-Cell Controller to capture 5 000-8 000 single cells per reaction.Reverse transcription (first-strand cDNA generation), fulllength cDNA amplification, and 3’ library construction were conducted using a Chromium Single-Cell 3’ Reagent Kit v3(10X Genomics, USA) following the manufacturer’s user guide. The libraries were sequenced on the Illumina NovaSeq sequencing platform with a paired-end read length of 150 bp.
Data processing and quality control
Raw sequencing data were processed using Cell Ranger(v3.1.0, 10X Genomics) with default mapping parameters,except for the use of the pre-mRNA version of theM. mulattareference genome from Ensembl, which was customized under 10X Genomics official instructions to ensure the capture of intronic reads originating from pre-mRNA transcripts abundant in the nuclear fraction. The feature-barcode matrices generated by Cell Ranger were loaded into the subsequent analysis pipeline.
Cells that passed the following filtration were retained for downstream analysis: number of detected genes per nucleus at least 500, except for some cerebral cortex samples in which the number of detected genes had to be at least 1 500 based on iterative analysis results; unique molecular identifier (UMI)counts per nucleus less than 50 000; percentage of UMIs from mitochondrial genes below 15%; doublet score calculated by Scrublet package (Wolock et al., 2019) below 0.25.
Dimension reduction and clustering
The Scanpy (v1.4.5.post3) package (Wolf et al., 2018) was used to perform preprocessing of gene expression data. The filtered gene-cell matrix was normalized to 104molecules/cell for sequencing depth with the “normalize_total” function and log transformed with the “log1p” function. Data variation caused by the number of UMI counts and percentage of UMIs from mitochondrial genes was regressed out using the“regress_out” function. Variable genes were obtained with mean expression levels ranging from 0.0125 to 3 and dispersion greater than 0.5. Uniform manifold approximation and projection (UMAP) was performed with the first 30 principal components obtained from principal component analysis (PCA) (batch effect corrected by Harmony(Korsunsky et al., 2019)) for visualization of single cells. Cell clustering was performed using the Leiden algorithm.
Analysis of differentially expressed genes (DEGs)
Differential gene expression analysis of each cell type between infected and healthy samples was performed using the “FindMarkers” function in the Seurat package (Butler et al.,2018) in R. The “MAST” method was applied. We only tested those genes detected in at least 10% of either healthy or infected samples for each cell type. We used “adjustedPvalue (Bonferroni corrected) <0.05” as a threshold to filter genes. Log2FC was calculated and used to rank the DEGs.
To identify marker genes for each cell group and facilitate the construction of dot plots of marker genes for each cell type, the “FindAllMarkers” function in the Seurat package was adopted.
Gene Ontology (GO) term and pathway enrichment analysis of DEGs
Enrichment analysis of GO terms (Biological Process) and pathways for infection-associated DEGs (|log2FC|>0.25,adjustedP<0.05) was implemented using Enrichr (Chen et al.,2013; Kuleshov et al., 2016) (https://maayanlab.cloud/Enrichr/). GO terms and pathways were considered significantly enriched atP<0.05.
Significance tests for selected genes
We performed the one-sided Wilcoxon Rank-Sum test to compare differences in gene expression between infected and healthy samples based on log-normalized expression data.Results were considered statistically significant at adjustedP<0.05.
Subclustering of astrocytes
Astrocyte subclustering was performed using scaled data with 4 000 highly variable genes with the Seurat package (Butler et al., 2018) in R. The “RunPCA” function was used for PCA to reduce dimensionality. Nearest neighbors were computed using the “FindNeighbors” function. Groups were clustered based on the Louvain algorithm using the “FindClusters”function. The “RunUMAP” function was used for visualization.
Trajectory analysis
We performed pseudotime trajectory analysis using the R package Monocle3 (Cao et al., 2019) to study cell state changes in astrocytes in the cerebral cortex. We first imported the data derived from Seurat into Monocle3, including an input read count matrix with the top variable features selected by the “FindVariableFeatures” function and a UMAP layout.Trajectory was then fitted using the “learn_graph” function and cells were ordered using the “order_cells” function in Monocle3.
Calculating module score
We applied the “AddModuleScore” function in the Seurat package to investigate differences between infected and healthy samples in terms of astrocyte reactivity and microglial activation in the cerebral cortex and necrotic cell death in the lung and kidney. One-sided Wilcoxon rank-sum test was used to check whether infected samples scored higher than healthy controls. Tests with adjustedP<0.05 were considered significant.
Analysis of regulons
We performed SCENIC analysis (Aibar et al., 2017) to study gene regulons in astrocytes using the “pySCENIC” package in Python. We used count data as input and applied the standard SCENIC workflow (grnboost2 - cistarget - AUCell). After obtaining the regulon activity matrix, we used “MAST” to identify differential regulons between infected and healthy samples of astrocytes. Regulons withP<0.05 were filtered out.
Cell-cell communication analysis
CellChat (v1.1.3) (Jin et al., 2021) was used to explore changes in cell-cell communication among cell types in lung tissue, with the aim to determine: (1) whether cell-cell communication is enhanced in the infected group compared with the healthy group; (2) which cell-type interactions are significantly altered by infected status; and (3) which signaling pathways mediate changes in communication. First, DEGs were identified for all cell groups between the infected and healthy groups; second, ensemble average expression was calculated for each cell group; third, intercellular communication probability was calculated based on the projection of ligand-receptor expression profiling onto proteinprotein networks; last, significant differences in intercellular communication between the infected and healthy groups were identified by a permutation test. The probability of communication at the signaling pathway level was inferred by calculating the probability of communication for all ligandreceptor interactions associated with each signaling pathway.Several signaling pathways were highlighted by ranking signaling from the infected and healthy groups. Cell group and ligand-receptor contributions of these pathways were further computed and visualized. Calculations were performed using the default parameters following the CellChat workflow.
CellPhoneDB software (Efremova et al., 2020) was used to determine cell-cell communication patterns between cell types in the infected samples of the cerebral cortex. This method mainly evaluates the number of ligand-receptor interactions of every two cell types. Interaction pairs were regarded as significant atP<0.05.
Immunofluorescence staining
Formalin fixed paraffin-embedded (FFPE) tissue sections(5 μm) were dewaxed in xylene, then rehydrated in 100%,95%, 85%, 75%, and 50% gradient ethanol and distilled water.Heat antigen retrieval was performed by steaming at 98 °C in retrieval solution (10 mmol/L sodium citrate, pH=6.0) for 20 min. Sections were allowed to cool at room temperature.Following antigen retrieval, sections were permeabilized in PBS containing 0.1% Triton X-100, then blocked using 5%-10% donkey serum in PBS for 1 h. The cerebral cortex sections were incubated with a primary antibody (rabbit anti-GFAP, Abcam (UK), ab7260, 1:500) overnight at 4 °C. A secondary antibody (donkey anti-rabbit, Alexa Fluor™ 488,Invitrogen (USA)) diluted in blocking buffer (1:500) was applied for 2 h at room temperature. For staining of macaque immunoglobulin G (IgG), rabbit anti-monkey IgG H&L (Alexa Fluor® 488) antibody (Bioss (China), bs-0335R-AF488, 1:500)was incubated with blocked kidney sections for 2 h at room temperature. Cell nuclei from all sections were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, Invitrogen (USA),D1306). Immunostaining images were acquired using an Olympus laser confocal microscope (FV3000) and analyzed with Olympus confocal software and Adobe Photoshop CC 2018.
RESULTS
Integrative analysis of single-nucleus transcriptomes of multiple macaque organs
We performed single-nucleus RNA-sequencing (snRNA-seq)of lung, liver, kidney, and cerebral prefrontal cortex tissues obtained from two young rhesus macaques (3 years old, one male, one female) infected with SARS-CoV-2 strain 107 (as described in our previous studies (Song et al., 2021; Song et al., 2020)) and sacrificed at 7 dpi (Figure 1A). Viral loads in the lung tissue were detected by qRT-PCR, which confirmed successful infection (Supplementary Figure S1 and Table S1).We performed snRNA-seq using samples of the same organs taken from healthy young macaques as controls (two healthy macaques for cerebral cortex and one macaque for the other three organs) (Figure 1A). After quality control and batch effect correction, we obtained a total of 77 029 nuclei from all four organs (lung,n=22 141 nuclei; liver,n=17 732 nuclei;kidney,n=13 550 nuclei; cerebral cortex,n=23 606 nuclei)(Supplementary Figure S2 and Table S2). Cell types were annotated with classical marker genes in each organ(Supplementary Figure S3 and Table S3). Integrated data across all four organs were visualized using UMAP(Figure 1B). Alveolar epithelial cells in the lung, hepatocytes in the liver, tubular cells in the kidney, and neural cells in the cerebral cortex were the exclusive parenchymal cell types identified in the respective organs. Certain cell types were largely mixed and similar in the four organs, e.g., endothelial cells, stromal cells, and immune cells. The liver exhibited the greatest transcriptional disturbance among organs, as indicated by the number of DEGs identified at the all-cell level(Figure 1C; Supplementary Table S4), which may be the result of the diverse metabolic functions of the liver.
Five DEGs were shared among the DEGs in the lung, liver,and kidney. The expression levels of two glycosyltransferasecoding genes, fucosyltransferaseFUT8,which is the sole enzyme catalyzing core fucosylation and is required for T cell activation (Liang et al., 2018), and sialyltransferaseST6GAL1,which enhances B cell-mediated humoral immunity (Irons et al., 2020), were down-regulated in the three aforementioned visceral organs at the all-cell level (Figure 1D), suggesting that adaptive immune responses are likely suppressed during SARS-CoV-2 infection. In addition,DNMT1, which encodes DNA methyltransferase 1, was shared by all DEG datasets from the four organs (Figure 1D). Transcriptional regulation is largely ruled by epigenetic mechanisms, such as DNA methylation (Zhu et al., 2016), hence the universal dysregulation ofDNMT1in various organs may underlie the infection phenotype of broadly altered transcriptional programs. Therefore, changes in the epigenetic landscape in host cells following infection may indicate a molecular mechanism used by SARS-CoV-2 to perturb host defense programs and physiological functions.
SARS-CoV-2-infected macaque lungs showed epithelial injury and fibrotic signatures similar to those in human COVID-19 patients
In the lung dataset, alveolar type I (AT1) and type II epithelial cells (AT2) were the most abundant cell types, with other epithelial cells, such as ciliated and club cells, also recovered(Figure 2A-C). The cell types most affected by SARS-CoV-2 infection included AT2, AT1, ciliated cells, endothelial cells,fibroblasts, and interstitial macrophages (Figure 2D;Supplementary Table S5). Previous studies have demonstrated that SARS-CoV-2 uses angiotensin converting enzyme 2 (ACE2) and transmembrane serine protease 2(TMPRSS2) as the cell-surface receptor and cellular protease for spike protein binding and priming, respectively (Hoffmann et al., 2020; Scudellari, 2021; Zhou et al., 2020). Therefore,we measured the coexpression of these two well-known SARS-CoV-2 entry factors (percentage of positive cells with read counts >0 for both genes) and found enrichment in the AT2 cells (Figure 2E), consistent with previous findings in human and cynomolgus monkeys (Ma et al., 2021; Muus et al., 2021; Sungnak et al., 2020). The proportion of AT2 cells was markedly reduced in infected samples compared to the healthy control (Figure 2C), in line with previous single-cell transcriptomic research using autopsy tissue samples from COVID-19 patients (Delorey et al., 2021; Melms et al., 2021),likely reflecting direct infection-induced apoptosis or cell death as AT2 cells possess entry factor expression. Accordingly, GO(Biological Process) and pathway enrichment analysis of DEGs in the AT2 cells revealed up-regulation of “positive regulation of programmed cell death” and “apoptosis”(Figure 2F). For instance,APAF1,BCLAF1,TNFRSF10A,TNFRSF21, and caspase-10 (CASP10), which participate in apoptotic induction, were up-regulated in AT2 cells(Supplementary Figure S4A). In addition, RNA metabolic processes were significantly enriched in up-regulated genes in AT2 cells (Figure 2F). For example, heterogeneous nuclear ribonucleoproteins (HNRNPs), representing a large family of factors responsible for RNA processing, including RNA splicing, maturation, decay, and translation, were up-regulated in AT2 cells (Supplementary Figure S4A). “Bacterial invasion of epithelial cells” and “endocytosis” also implied direct infection of AT2 cells. Together, these results suggest that AT2 is the potential lung cell type targeted by SARS-CoV-2.The maintenance of alveolar epithelium homeostasis and its regeneration after injury are fueled by surfactant-producing AT2 cells, which can self-renew and differentiate into AT1 cells. Regeneration-related genes critical for lung injury repair were down-regulated in AT2 cells after SARS-CoV-2 infection (Supplementary Figure S4B), suggesting impairment in the differentiation ability of AT2 cells, consistent with that found in COVID-19 patient lungs (Melms et al., 2021; Wang et al.,2021).
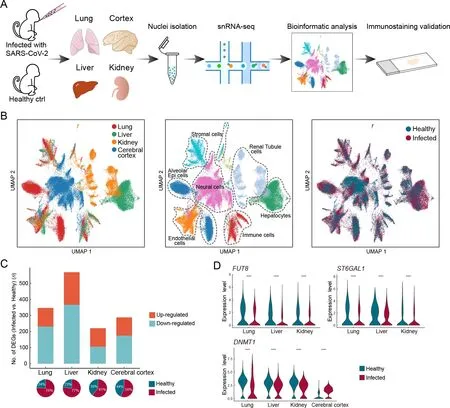
Figure 1 Overview of single-nucleus transcriptomes from four organs in rhesus macaques

Figure 2 Epithelial cell impairment and signaling pathway changes in macaque lungs after SARS-CoV-2 infection
The dysregulated biological processes of AT1 cells showed similarities to those of AT2 cells. Notably, cytokine-associated pathways were elevated in AT1 cells (Supplementary Figure S4C), suggesting activation of the inflammatory response in the lung after infection.LMOD1, a marker gene of idiopathic pulmonary fibrosis (Selman et al., 2006), was one of the most up-regulated genes in the AT1 cells (log2FC=2.03). In addition, collagen-related genes were highly expressed in fibroblasts from infected samples (Supplementary Figure S4D), suggesting a contribution to fibrosis. Orphan nuclear receptorsNR4A1andNR4A2,which inhibit fibrosis (Chen et al., 2015; Palumbo-Zerr et al., 2015), were significantly down-regulated (Supplementary Figure S4E). Moreover,fibrogenic factorTIMP2and protective antifibrotic factorTIMP3(Kassiri et al., 2009; Nie et al., 2004) were both dysregulated in the profibrotic direction (Supplementary Figure S4E). These observations suggest the occurrence of fibrosis in the lungs of SARS-CoV-2-infected macaques.
Ciliated cells exhibited impaired physiological functions associated with cilia beating and mucus removal in the SARSCoV-2-infected lungs. Numerous genes closely associated with cilium formation and movement were down-regulated(Figure 2G), consistent with previous study of bronchoalveolar lavage fluid (BALF) in COVID-19 patients (He et al., 2020).Consistent with the reduction in ciliated cell proportion(Figure 2C), the necrotic cell death score was significantly higher in ciliated cells from infected samples than from the healthy control, while other cell types were minimally changed by infection (Figure 2H). CD47, a cell-survival signaling molecule that interacts with SIRPα (Lehrman et al., 2018;Okazawa et al., 2005), showed significantly higher expression in ciliated cells from infected samples than from the healthy control (Figure 2G), possibly due to the preferential survival of cells with elevated CD47 expression. These observations suggest that ciliated cells are acutely affected by SARS-CoV-2 infection, leading to necrosis.
Antigen presentation deficiency and signaling pathway alterations in macaque lungs
Although macrophages are professional antigen-presenting cells (APCs), the expression levels of major histocompatibility complex (MHC) component genesHLA-DRA,HLA-DRB1,HLA-DPA1,HLA-C,HLA-B, and chaperone moleculeCD74were surprisingly down-regulated in the lung macrophages after SARS-CoV-2 infection (Supplementary Figure S4F-H).Moreover, we observed reduced expression of MHC class I/II genes in non-classical APCs, including alveolar epithelial cells and endothelial cells (Supplementary Figure S4H). Therefore,we propose that suppression of MHC class I/II-related components responsible for antigen presentation and adaptive immunity may be a potential immune evasion strategy for SARS-CoV-2 to target the MHC system.
To identify changes in global cellular communication in the lung after infection, we used the intercellular communication inference tool “CellChat” (Jin et al., 2021). We found that angiopoietin (ANGPT) and fibroblast growth factor (FGF)signal intensities were significantly increased in infected samples (Supplementary Figure S5A). The ANGPT signaling pathway was activated in sender cell types other than fibroblasts and pericytes, and in receiver cell types other than endothelial cells (Figure 2I; Supplementary Figure S5B). The ANGPT1-TEK pair became more dominant in infected samples compared to the control (Figure 2I). This pair maintains the endothelial barrier and blood vessel integrity and plays a role in anti-inflammation (David et al., 2013;Huang et al., 2010). Therefore, an increase in their signaling may facilitate tissue injury repair after infection. In addition, the FGF signaling pathway showed increased fibroblast and mesothelial cell activity in the lung (Figure 2J; Supplementary Figure S5C). Among the most frequent ligand-receptor pairs,the contribution of FGF7-FGFR2 to FGF signaling was substantially increased after SARS-CoV-2 infection(Figure 2J). FGF7 is thought to regulate pulmonary epithelial proliferation and facilitate lung repair (Xie et al., 2020; Yano et al., 2000). These results suggest that the spontaneous lung injury repair program was initiated in macaques by 7 dpi or earlier.
Inflammatory response and immune tolerance in macaque liver
Hepatocytes, which were the predominant cells in the liver(Figure 3A-C), were enriched inACE2andTMPRSS2coexpression, especially zone 1 hepatocytes (Figure 3D), and contained the most DEGs between infected and healthy livers(Figure 3E; Supplementary Table S6). Although the proportion of coexpression was notably higher in the healthy control cholangiocytes, the proportion in infected samples was zero(Figure 3D), which may be due to the extremely low number of cholangiocytes captured in infected samples (Figure 3C).Therefore, we inferred that hepatocytes may be the potential target cell type in the liver and preferentially affected during SARS-CoV-2 infection.
DEG enrichment analysis in hepatocytes indicated that the complement system was activated in infected samples, as evidenced by the activation of various pathways, including the classical, alternative, and lectin-induced complement pathways (Figure 3F). The substantial increase in complement component proteins C5, C6, C8A, and C9 (log2FC=1.94, 1.44,1.26, and 0.97, respectively) may result in enhanced formation of membrane attack complex (MAC), which is incorporated into the cell membrane to cause cell lysis. “Response to cytokine” was also enriched in the up-regulated genes of hepatocytes (Figure 3F). Cytokine receptorsIL1R1,IL1R2,IFNAR2, andIFNGR2were elevated, as wereIRAK2andIL6ST, which are regulators and signal transducers that participate in cytokine-induced activation of the JAK-STAT3 and NF-κB signaling pathways. Accordingly, the expression levels ofSTAT3,NFKB1,NFKB2,RELB, andRELwere also up-regulated (Figure 3G). Therefore, after SARS-CoV-2 infection, innate immune defense-complement system and inflammatory response were activated in the macaque livers,especially in the hepatocytes.
The liver has multiple metabolic functions in the body.Various metabolic processes, including those involving lipids, lipoproteins, fatty acids, glucose, and glycogen, were dysregulated in the liver after SARS-CoV-2 infection(Figure 3H; Supplementary Figure S6A). Bile is a critical aqueous liver secretion produced by hepatocytes that facilitates dietary fat absorption in the intestinal lumen. Here,bile biosynthesis and secretion-related processes were significantly enriched in down-regulated genes in hepatocytes(Figure 3H). For example, the expression levels of genes encoding membrane transporters involved in bile secretion,such asABCB1,ABCC3,ABCC6,SLC22A1, andSLC22A7(Boyer, 2013), were decreased in the hepatocytes (Figure 3I),indicating that bile generation and secretion may be impaired in the liver after SARS-CoV-2 infection.

Figure 3 Hepatocyte dysregulations and immune evasion pathway in the liver of SARS-CoV-2-infected macaques
The liver also plays a central role in the blood coagulation cascade as it produces most coagulation factors. In this study,we found extensive dysregulation of coagulation factors,inhibitors, and fibrinolysis-associated proteins in the liver after infection (Figure 3J), especially in hepatocytes and endothelial cells, which are the primary sources of these coagulation factors. TheF2gene encoding prothrombin, the active form(thrombin) of which plays a central role in hemostasis(Crawley et al., 2007) by converting fibrinogen into fibrin to form blood clots, was down-regulated. ThePLGgene encoding plasminogen (Schuster et al., 2007), the active form(plasmin) of which plays a key role in fibrinolysis by dissolving fibrin in blood clots, was up-regulated (Figure 3J). These observations suggest that SARS-CoV-2 infection may lead to a higher risk of bleeding.
The liver is the largest solid organ in the body and contains many resident immune cells. However, we found no activation signatures of macrophages or T cells, other than up-regulation of complement components (Figure 3K, L). Surprisingly,tryptophan metabolism along the kynurenine (Kyn) pathway was activated in the liver following SARS-CoV-2 infection. The Kyn pathway is a key mechanism for creating an immunosuppressive microenvironment, which facilitates immune surveillance escape by many types of cancer cells(Adams et al., 2012; Liu et al., 2020b; Nevler et al., 2019; Rad Pour et al., 2019). Activation of the Kyn pathway has also been reported in viral infection (Jenabian et al., 2015; Ma et al., 2009; Pizzini et al., 2019).
Indoleamine 2,3-dioxygenase 2 (IDO2) and tryptophan 2,3-dioxygenase (TDO2), which catabolize the substrate tryptophan to form Kyn, are first and rate-limiting enzymes of the Kyn pathway. Here, the expression levels ofIDO2andTDO2were elevated in hepatocytes (Figure 3F), andIDO2was also elevated in macrophages and T cells after SARSCoV-2 infection (Figure 3K, L). Moreover, Kyn 3-monooxygenase (KMO), which catalyzes the hydroxylation of Kyn to form 3-hydroxykynurenine (3-HK), and Kyn aminotransferase 3 (KYAT3), which catalyzes the transamination of Kyn to form kynurenic acid (KynA), were upregulated at the transcriptional level in hepatocytes(Figure 3F).KMOgene expression was also elevated in immune cells (Figure 3K, L). Tryptophan is an amino acid required for the survival and proliferation of T cells, such as T helper (Th) cells and cytotoxic T lymphocytes (CTLs)(Fallarino et al., 2002; Lee et al., 2002). Thus, our results suggest that immune cell populations are suppressed in the tryptophan-deprived microenvironment established by the overexpression ofIDO2andTDO2after SARS-CoV-2 infection.
Importantly, the aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor, which can be activated by binding to endogenous tryptophan derivatives including Kyn and KynA (Dinatale et al., 2010; Opitz et al., 2011). The Kyn-AHR signaling pathway suppresses the differentiation and activity of immune cells, resulting in an impaired antitumor immune response (Liu et al., 2018; Mezrich et al., 2010; Opitz et al., 2011). Along with Kyn accumulation resulting from increased dioxygenases,AHRgene expression was upregulated in hepatocytes (Figure 3F), macrophages(Figure 3K), and T cells (although not significantly in T cells).Consistent with the immunosuppressive effect described above, the proportions of macrophages and T cells were reduced in the infected liver samples (Figure 3C), and genes associated with apoptosis, includingCRADD,GAS2,FHIT,andZNF385B, were up-regulated in these immune cells(Figure 3K, L).
The Kyn pathway is induced by proinflammatory cytokines(Brooks et al., 2016; Dostal et al., 2018; Santhanam et al.,2016). As described previously, cytokine receptor genes,signal transducer genes, NF-κB subunit genes, and cytokine and chemokine genes, such asIL1A,IL6,CXCL2, andCXCL10, were up-regulated in the liver (Figure 3G, K, L;Supplementary Figure S6B). Therefore, given the integrated activation of the Kyn pathway and its immunoregulatory effects (Wu et al., 2018), as well as the inflammatory signatures observed in the liver, we propose that inflammation triggered by SARS-CoV-2 infection induced Kyn pathway activation, leading to the establishment of an immunosuppressive microenvironment and immune tolerance in the macaque liver. These mechanisms are summarized in Figure 3M, providing new insights into liver manifestations and novel immunomodulatory therapies for SARS-CoV-2 infection.
Renal tubule tropism and NELL1-associated membranous nephropathy in the kidney
The kidney is a selective filtering organ composed of capillary vessels and renal tubules divided into segments, including proximal tubules, loops of Henle (LoH), distal convoluted tubules, connecting tubules, and collecting ducts (Figure 4A).Here, the proportion of each parenchymal cell type showed minimal change after infection (Figure 4B, C). Coexpression ofACE2andTMPRSS2was highlighted in the proximal tubule cells (Figure 4D), indicating that proximal tubules may be more vulnerable to SARS-COV-2 infection, consistent with findings reported in human kidney cells (Pan et al., 2020).Accordingly, proximal tubule cells were the most severely affected cell type, with more DEGs than any other cell type(Figure 4E; Supplementary Table S7). Based on the necrotic cell death scores for all parenchymal cell types, the necrosis score was significantly higher for proximal tubule cells in the infection group than in the healthy control (Figure 4F),suggesting the formation of necrotic lesions in the proximal tubules of infected macaques, in accordance with the renal histopathological features of proximal tubule acute necrotic injury and intraluminal necrotic epithelium debris found in autopsy samples of COVID-19 patients (Su et al., 2020;Werion et al., 2020).
Our results also indicated that LoH cells may be affected, as up-regulated genes in the “LoH3” cluster were enriched in pathways related to the viral life cycle, including “viral release”,“exocytosis”, “vesicle-mediated transport”, “viral transcription”,and “endocytosis” (Figure 4G). Specifically, CLTA and CLINT1 are involved in the formation and trafficking of clathrin-coated vesicles and are reportedly associated with SARS-CoV-2 entry via endocytosis (Bayati et al., 2021). RAB7A is an important regulator of vesicular transport and endolysosomal trafficking and is required for vesicular trafficking and cell surface expression of ACE2 (Daniloski et al., 2021). GOLGA4 plays a key role in vesicular transport at the Golgi apparatus level and is also involved in endosome-to-Golgi trafficking.VPS4A functions in membrane fission events, such as cytokinesis and enveloped virus budding (Chen & Lamb,2008), and RIMS2 and EXPH5 are effector proteins involved in exocytosis. Elevated expression of these viral-life-cycle molecules (Figure 4H) suggests that SARS-CoV-2 likely infected LoH3 cluster cells, where virions underwent internalization, transport, and release. Thus, in addition to the vulnerable proximal tubule in the renal cortex, the LoH in the renal medulla may also be a target of SARS-CoV-2, which deserves further study.
However, the LoH3 cluster did not contain a high percentage ofACE2-andTMPRSS2-coexpressing cells(Figure 4D). Interestingly, we assessed the expression of several potential coronavirus entry factors (Singh et al., 2020)in LoH3 cells (Supplementary Figure S7A) and found that theNRP1gene was significantly increased in these cells of the infected macaques. The protein encoded byNRP1is a cofactor that can facilitate and potentiate SARS-CoV-2 infectivity (Cantuti-Castelvetri et al., 2020). In addition,CTSL,a substitutive protease that primes the spike protein of SARSCoV and possibly SARS-CoV-2 (Simmons et al., 2013), was markedly up-regulated in LoH3 cells of infected samples.NRP1andCTSLwere also up-regulated in the proximal tubules (Supplementary Figure S7B). Thus, these results suggest that the increase inNRP1andCTSLexpression following infection may be a positive-feedback molecular mechanism for SARS-CoV-2 entry into the kidney.
Podocytes in the renal corpuscle form the final filtration barrier of the glomerulus and play a critical role in preventing the loss of protein in urine. Podocyte dysfunction can easily induce proteinuria, a common manifestation in COVID-19 patients (Pei et al., 2020). Therefore, we speculated that podocyte dysregulation may also occur in infected macaques.Notably,NELL1(encoding neural epidermal growth factor-like 1) expression was specifically and highly up-regulated in podocytes of the infection group (log2FC=5.8) (Figure 4I).NELL1 is a novel antigen marker for membranous nephropathy (MN), in addition to the well-studied and common antigen marker PLA2R (Caza et al., 2021; Sethi et al., 2020).In NELL1-associated MN, NELL1 is shed from podocytes and binds to autoantibodies against NELL1 to form antigenantibody complexes localized in the glomerular basement membrane. Here, immunostaining of macaque IgG in kidney sections showed segmental positive IgG staining in glomeruli of infected samples (Figure 4K), while IgG staining was negative in the healthy samples (Figure 4J). This finding suggests that SARS-CoV-2 infection may lead to the development of NELL1-associated MN in macaques. This discovery provides new insights into SARS-CoV-2-associated kidney disease, warranting further attention in the clinical treatment of COVID-19 patients.
Astrocyte reactivity and myelination defects in macaque cerebral cortex
Various neuronal and glial cell types were captured in our cerebral cortex atlas (Figure 5A-C), with astrocytes containing the most DEGs (Figure 5D; Supplementary Table S8). We found that astrocyte reactivity marker genes, includingSTAT3,GFAP,NFIA,MT1M,NFATC1,S100B, andMAOB(Escartin et al., 2021; Laug et al., 2019), were significantly up-regulated in astrocytes (Figure 5E), indicating that astrocytes were in a reactive state in the infected macaques. Reactive astrogliosis may explain the marked increase in the proportion of astrocytes in the infected samples (Figure 5C). Under physiological conditions, in addition to participating in bloodbrain barrier formation with endothelial cells and pericytes,astrocytes regulate neuronal activities via tripartite synapses.Specifically, astrocytes subtly regulate the levels of the excitatory neurotransmitter glutamate in the synaptic cleft via glutamate transporters EAAT1 (SLC1A3) and EAAT2(SLC1A2) located in the astrocyte end-feet. The expression levels ofSLC1A3andSLC1A2were substantially upregulated in the astrocytes (Figure 5F), which may lead to lower glutamate levels and reduced neuronal excitability.

Figure 4 Tubule cell tropism and glomerular involvement in the kidney of SARS-CoV-2-infected macaques
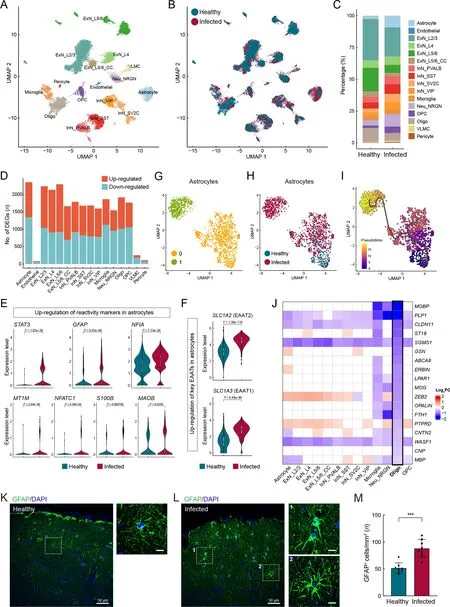
Figure 5 Pathological states of astrocytes and oligodendrocytes in cerebral cortex of SARS-CoV-2-infected macaques
We further investigated astrocytes using unsupervised clustering and identified two major subclusters (Figure 5G).The two subclusters exhibited relatively even distributions in terms of sample and gender, as shown in UMAP (Figure 6A,B), indicating minimal technical and/or batch artifacts.Subcluster 1 was an infected sample-specific subpopulation,predominantly composed of cells from infected samples(Figure 5H). The astrocyte reactivity score, calculated based on the expression levels of the seven reactive marker genes mentioned earlier, was significantly higher in infected samplespecific subcluster 1 than in other astrocytes (Figure 6C). The expression levels ofSLC1A2andSLC1A3were also significantly higher in subcluster 1 than in other astrocytes(Figure 6D). Trajectory analysis revealed that infectionassociated subcluster 1 emerged from the parental homeostatic population (Figure 5I), further suggesting that these astrocytes emerged in response to the pathological environment.
Hypertrophy of cellular processes is a typical phenotype of reactive astrocytes (Pekny & Pekna, 2014). Here, GFAP immunostaining of prefrontal cortex sections to label astrocyte morphology showed evident signs of characteristic hypertrophy in infected macaques (Figure 5K, L). The number of GFAP-positive astrocytes in infected animals was significantly higher than that in the healthy macaques(Figure 5M), further validating reactive astrogliosis. These findings suggest the occurrence of astrocyte reactivity and dysregulation of excitatory synaptic transmission in SARSCoV-2-infected macaques, with these perturbations in astrocytes inducing an infected sample-specific subpopulation at the molecular level.
We next used CellPhoneDB (Efremova et al., 2020) to infer the potential cell-cell communication network mediated by ligand-receptor interaction (LRI) pairs in infected cerebral cortex samples (Figure 6E). Here, we focused on communication between astrocyte subcluster 1 and other cell types to investigate the possible underlying dysregulation.Among the identified significant LRI pairs, we found that the NRG1-ERBB4 interaction pair between astrocyte subcluster 1 and excitatory neurons was quite relevant (Figure 6F). As NRG1-ERBB4 signaling is implicated in psychiatric disorders,including epilepsy (Zhu et al., 2017) and schizophrenia(Banerjee et al., 2010), the increase in NRG1-ERBB4 signaling triggered by the infected sample-enriched astrocyte subcluster may cause psychiatric symptoms in SARS-CoV-2-infected macaques, although this was not tested here. In addition, we identified two highly manifested regulons in astrocytes from the infected samples (Figure 6G).SLC1A2andSLC1A3were predicted to be regulated byTCF7L2(Figure 6G),one of the top DEGs identified in the astrocytes(log2FC=2.2).TCF7L2can also activateIDO1expression(Figure 6G), which may lead to more activated tryptophan metabolism in the brain, as revealed in the liver, and may explain the lack of activation signatures in microglia(Figure 6H).
Oligodendrocytes in the CNS are responsible for producing the myelin sheath that insulates axons. Common markers for oligodendrocytes includeMOBP,MOG,MBP, andPLP1,which encode structural components of the myelin sheath and play important roles in myelin sheath completion and maintenance. TheCLDN11gene encodes claudin 11, which is specific to oligodendrocytes and facilitates the formation of tight junctions in the myelin paranodal loops (Morita et al.,1999). The actin disassembly protein gelsolin encoded byGSNpromotes actin disassembly and is required for myelin wrapping, with high enrichment in actively myelinating oligodendrocytes (Zuchero et al., 2015). The Erbb2 interacting protein (ERBIN) is necessary for remyelination of regenerated axons after injury (Liang et al., 2012).PTPRDis expressed in oligodendrocytes at postnatal stages and contributes to the initiation of axonal myelination (Zhu et al., 2015). The cell adhesion molecule encoded by theCNTN2gene can affect the expression levels of myelin and myelin-regulating genes and is also responsible for oligodendrocyte branching (Zoupi et al., 2018). TheCNPgene encodes a phosphodiesterase that interacts with tubulin and promotes microtubule assembly and is thus necessary for oligodendrocyte process outgrowth and arborization (Lee et al., 2005). Intriguingly, we found that many genes closely associated with myelin, including those mentioned above, were specifically down-regulated in oligodendrocytes (Figure 5J), predicting likely myelination defects in the cerebral cortex of infected macaques. As myelin sheaths around neuronal axons enable rapid impulse propagation of electrical signals in the CNS, the hypomyelination or demyelination in the cerebrum of infected animals may impair axonal conduction. Overall, SARS-CoV-2 infection-associated perturbations in the macaque cerebral cortex, including pathological astrocytes and oligodendrocytes, and subsequent neuronal impairment of synaptic excitability transmission and axonal conduction were evident, and may contribute to cognitive impairment and psychiatric symptoms after infection (Figure 6I).
Endothelial cell dysfunction and circadian rhythm disruption after infection
Endothelial cells are highly involved in COVID-19 pathologies(Varga et al., 2020). Thus, we performed integrative analysis of endothelial cells from the four tested organs and found different cellular perturbations based on organ-specific physiological functions (Figure 7A). Expression of ACE,responsible for converting angiotensin I into angiotensin II and leading to increased vasoconstriction and inflammation(Givertz, 2001), was specifically decreased in the lung endothelial cells (Figure 7A), suggesting a vasoprotective effect, consistent with the observed tendency toward lung injury repair after several days of exposure to SARS-CoV-2.MHC components, including human leukocyte antigen (HLA)class I/II genes, were down-regulated in the lung endothelium(Figure 7A), further indicating that inhibition of the MHC pathway may be an immune evasion strategy used by SARSCoV-2.
Endothelial cells in the liver showed unique dysregulation of Rho GTPase regulators, including inhibitor GAPs and activator GEFs (including DOCKs) (Figure 7A), which play key roles in regulating vascular barrier function and integrity via the actin cytoskeleton (Beckers et al., 2010), potentially leading to impaired endothelial barrier function in the liver after infection.PODXL plays a role in orchestrating interactions with extracellular matrix components and basement membranes to regulate vascular integrity and permeability (Debruin et al.,2014). The levels of PODXL and tight junction protein CLDN2,as well as basement membrane laminin components LAMB1 and LAMB4, were specifically increased in the kidney endothelial cells (Figure 7A), possibly contributing to glomerular permeability disorder in the kidney, in addition to MN. Endothelial cells in the cerebral cortex exhibited several specific changes in potassium and calcium channels associated with nervous system functions (Figure 7A).
Endothelial cells in the different organs showed several shared transcriptional changes (Figure 7B). RIG-I is a key sensor for recognizing nucleic acids derived from RNA viruses, andSEC14L1encodes a negative regulator of the RIG-I antiviral signal (Li et al., 2013), and its down-regulation may help attenuate viral replication. Endomucin (EMCN)prevents leukocyte-endothelial cell adhesion, and thus reduced endomucin is critical to facilitate the adhesion of leukocytes to inflamed tissues (Zahr et al., 2016).ARHGAP18,an essential inhibitor of inflammation development in the endothelium (Coleman et al., 2020), andPPP1R16B(also known as TIMAP), a positive regulator of endothelial barrier function (Csortos et al., 2008), were both down-regulated in the endothelial cells of infected macaques. These changes suggest that endothelial cells exhibited elevated innate antiviral immunity, enhanced leukocyte adherence and extravasation, and impaired barrier function to some extent.DNMT1gene expression was also dysregulated in the endothelial cells of all four organs (Figure 7B), consistent with the findings presented in Figure 1D.
Recent studies have revealed an increasing prevalence of anxiety and circadian rhythm disorders during the COVID-19 pandemic (Boiko et al., 2022; Deng et al., 2021). A follow-up study reported that COVID-19-impacted people still experience fatigue, sleep difficulties, and anxiety or depression six months after acute infection (Huang et al.,2021). Whether the virus disrupts circadian rhythm and affects sleep behavior needs to be explored. Here, we observed that the expression of core components of the circadian clock was extensively dysregulated in the four organs of the SARS-CoV-2-infected macaques (Figure 7C). The visceral organs showed a similar pattern of circadian clock disruption: thePER3gene was up-regulated and other components were down-regulated or changed negligibly. In contrast,PER3andPER2were down-regulated in all neural cell types in the cerebral cortex,and theCLOCKparalogNPAS2, which plays a central role in the regulation of circadian rhythm, was disturbed in multiple neural cell types in a discordant manner. Thus, the widespread dysregulation of circadian clock components in our dataset provides molecular evidence linking SARS-CoV-2 infection with disruption of the circadian rhythm.

Figure 6 Evaluation of astrocytes in infected samples and summary of cerebral cortex perturbation
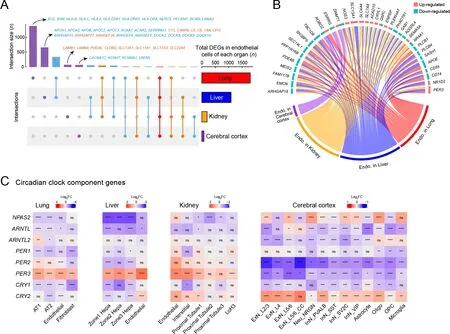
Figure 7 Endothelial cell dysfunction and circadian rhythm disruption in multiple organs of SARS-CoV-2 infected rhesus macaques
DISCUSSION
Previous studies have investigated the molecular basis of the pathological hallmarks of COVID-19 using postmortem specimens obtained from critically ill patients (Delorey et al.,2021; Melms et al., 2021; Wang et al., 2021). However,differences in age, underlying disease, and length of hospital stay and treatment of human subjects have contributed to the heterogeneity of COVID-19 samples and complicated the delineation of pathological mechanisms. Furthermore,responses of non-respiratory organs to SARS-CoV-2 infection are poorly understood at the single-cell level. In the present study, we explored single-cell transcriptomic changes in the lung, liver, kidney, and cerebrum in a rhesus macaque model at 7 dpi. Although we detected no viral reads via snRNA-seq,we observed notable cellular perturbations in all four organs,demonstrating multi-organ involvement in SARS-CoV-2 infection, further supporting the broad threat of the virus to the host body.
In terms of susceptibility, we identified AT2 cells,hepatocytes, and proximal tubule cells as potential target cells of SARS-CoV-2 according to the coexpression percentage ofACE2andTMPRSS2. A LoH cell subtype (cluster LoH3) was also identified as a probable target cell, as indicated by the upregulated expression of genes associated with the viral life cycle. In addition, ciliated cells and proximal tubule cells were severely impacted according to necrosis score analysis.Aberrant RNA metabolic processes in AT2 cells, such as upregulation of HNRNPs, also suggested that these cells may be a target in the lung. The influenza A virus utilizes hnRNP K to alter host splicing and promote infection (Thompson et al.,2018), and hnRNP A2/B1 interacts with NSP1 (non-structural protein 1) in SARS-CoV-2 to inhibit host interferon β (IFN-β)expression and facilitate β-coronavirus infection (Zhou et al.,2021). Thus, the up-regulated RNA metabolic processes observed in AT2 cells suggest that AT2 cells are infected, and mRNA processing-associated proteins are leveraged by SARS-CoV-2 to facilitate its transcription and replication.
In the cerebral cortex, we identified a marked increase in astrocyte reactivity and glutamate transporters. Furthermore,substantial down-regulation of myelin structural molecules and regulators pointed to myelination defects. Thus, neuronal perturbations may follow glial dysfunction, together causing neurological involvement. However, the expression level of acknowledged viral entry factors was extremely low in the cerebral cortex (data not shown), and thus the target cell type by which the virus enters the CNS remains unclear.
According to our multi-organ study, SARS-CoV-2 infection results in immune disorder. From a general view, reduced expression ofFUT8andST6GAL1was observed in the lung,liver, and kidney, as shown in Figure 1D, indicating that SARS-CoV-2 can restrain the host adaptive immune response to facilitate its own survival. From a multi-organ perspective,we found various adverse immune responses in the different organs.
In the lungs, we found decreased expression of MHC class I/II genes (mainlyHLA-C,HLA-DRB1, andCD74) in macrophages, epithelial cells, and endothelial cells after macaque infection, concordant with other studies of peripheral blood mononuclear cells (PBMCs) (Wilk et al., 2020), preenriched dendritic cell subsets from peripheral blood (Saichi et al., 2021), and nasopharyngeal swabs (Yoo et al., 2021)from SARS-CoV-2-infected patients. The suppression of MHC components in professional and non-classical APCs in the macaque lungs further confirmed that SARS-CoV-2 targets MHC-related pathways to suppress antigen presentation and facilitate immune evasion. In addition, the sole receptor for IFN-γ (IFNGR1) was down-regulated in macrophages, with IFN-γ known to be a primary regulator of MHC class I/II pathway activation (Goñalons et al., 1998; Zhou, 2009). Thus,we reasoned that the virus inhibits IFN-γ signaling, leading to insufficient MHC levels. Indeed, previous studies have demonstrated that SARS-CoV-2 can inhibit IFN signaling pathways (including types I, III, and II) in human patients(Blanco-Melo et al., 2020; Galani et al., 2021; Hadjadj et al.,2020; Yoo et al., 2021; Zhang et al., 2020), especially in severe cases, and an impaired IFN response is a driving feature of disease progression.
In addition to inhibiting antigen presentation in the lung, we also found that the Kyn pathway was nearly globally activated in the liver, as indicated by the up-regulation ofIDO2,TDO2,KMO, andAHRin hepatocytes and immune cells, including macrophages and T cells. In the Kyn pathway, key enzymes play significant roles in facilitating immune evasion in a variety of cancers, including hepatocarcinoma (Jin et al., 2015; Li et al., 2020). In addition to the potent immunosuppressive effects of IDO2/TDO2/KMO up-regulation-induced local tryptophan depletion and biologically active tryptophan catabolites, AHR activation by binding to the endogenous ligand Kyn suppresses T cell proliferation and function.Tryptophan catabolism has also been implicated in human immunodeficiency virus (HIV) (Jenabian et al., 2015), hepatitis B virus (HBV) (Ma et al., 2009), and influenza virus infections(Pizzini et al., 2019). Therefore, considering that the Kyn pathway was activated in the SARS-CoV-2-infected macaque livers, and given the immunomodulatory role of the Kyn pathway (Wu et al., 2018), we propose that SARS-CoV-2 infection can lead to immunosuppression or tolerance in the liver by manipulating the Kyn pathway.
Recent metabolomic analysis of blood plasma and serum based on liquid chromatography-mass spectrometry revealed reduced tryptophan levels and increased Kyn levels in the peripheral blood of COVID-19 patients (Lawler et al., 2021;Lionetto et al., 2021; Thomas et al., 2020). Our findings showing up-regulation ofIDO2andTDO2in the macaque livers further corroborated the activation of tryptophan metabolism along the Kyn pathway during SARS-CoV-2 infection. The immune evasion strategies of SARS-CoV-2 lead to a higher risk of prolonged infection and long-term symptoms. Therefore, it may be necessary for COVID-19-infected people and survivors to monitor their nutritional status, particularly levels of tryptophan and its metabolites.
Furthermore, immune disturbance caused by SARS-CoV-2 infection was also reflected in the discovery of MN in the kidney. MN is an autoimmune disease in which immune complexes inappropriately attack the filter membranes. PLA2R is the most common target antigen in primary MN (Ronco et al., 2021), while secondary MN can be secondary to infection, drugs, and malignancy. In our study, NELL1-associated MN was found in the kidney, and the activated complement system in the liver exacerbated MN by MAC(Cravedi, 2021). MN has rarely been reported in human COVID-19 infection (Guo et al., 2022) or vaccination (Aydin et al., 2021), with only a small fraction of these cases being PLA2R-positive. Hence, the observation of MN in our study provides new insights into SARS-CoV-2 infection-associated renal manifestations, although whether the MN in macaques is primary or secondary remains to be elucidated.
The current study has several limitations. First, we only included samples at 7 dpi for analysis. Longitudinal studies using samples collected at different times of infection and compared to infected groups after vaccination should help elucidate cell-type responses and gene expression patterns under such conditions. Second, although not performed in the current study, we will explore functional characterizations of the abovementioned genes to confirm their putative roles and test their potential as valid targets for treatment. We believe that the transcriptomic profiling landscape of single cells in different organs in this report should provide a solid basis for future studies.
Overall, this study provides evidence of multi-organ pathological changes in rhesus macaques following SARSCoV-2 infection, as well as novel findings not previously reported in human subjects. We identified potential target cell types and genes with dysregulated expression and enriched pathways. Our study supports rhesus macaques as useful models for studying SARS-CoV-2 pathology and highlights potential therapeutic targets for COVID-19. Future studies with larger sample sizes are expected to provide a more comprehensive understanding of the pathogenesis of SARSCoV-2 and host defense, particularly local adaptive immune responses in multiple organs.
DATA AVAILABILITY
The raw sequencing data used in this study were deposited in the Genome Sequence Archive (GSA) database of the National Genomics Data Center (NGDC) under accession number CRA006787, Gene Expression Omnibus (GEO) of the NCBI under accession number GSE217483, and Science Data Bank (doi:10.57760/sciencedb.06252). All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
X.W., Y.T.Z., and Y.G.Y. conceived the project and designed the experiments. T.Z.S., Z.H., J.B.H., L.X., and D.D.Y.constructed the animal model and collected the samples. Q.M.and Z.L. performed the single-nucleus RNA-seq experiments.W.M., Z.W., B.Z., and Y.S. analyzed the RNA-seq data. Q.M.and B.W. performed tissue sectioning and immunostaining.Q.M., W.M., and Q.W. wrote the manuscript and all authors edited and proofed the manuscript. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Dr. Changwen Ke (Guangdong Provincial Center for Disease Control and Prevention) for providing the SARS-CoV-2 strain.
杂志排行

Zoological Research的其它文章
- Unveiling the functional and evolutionary landscape of RNA editing in chicken using genomics and transcriptomics
- Antiviral effects of natural small molecules on aquatic rhabdovirus by interfering with early viral replication
- Virome in healthy pangolins reveals compatibility with multiple potentially zoonotic viruses
- Chronic lithium treatment ameliorates ketamineinduced mania-like behavior via the PI3K-AKT signaling pathway
- A glimpse into the biodiversity of insects in Yunnan: An updated and annotated checklist of butterflies(Lepidoptera, Papilionoidea)
- Dynamic coding in the hippocampus during navigation