Virome in healthy pangolins reveals compatibility with multiple potentially zoonotic viruses
2022-11-29FengJuanTianJingLiWenLiLiuYuJieLiuYunJiaHuQiHangTuYangLiYuBaiMangShiTengChengQueYanLingHuYiGangTong
Feng-Juan Tian, Jing Li, Wen-Li Liu, Yu-Jie Liu, Yun-Jia Hu, Qi-Hang Tu, Yang Li, Yu Bai, Mang Shi,Teng-Cheng Que, Yan-Ling Hu, Yi-Gang Tong,*
1 College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China
2 Centre for Infection and Immunity Studies, School of Medicine, Shenzhen Campus of Sun Yat-Sen University, Sun Yat-Sen University,Shenzhen, Guangdong 518107, China
3 Faculty of Data Science, City University of Macau, Macau 999078, China
4 Guangxi Zhuang Autonomous Terrestrial Wildlife Rescue Research and Epidemic Diseases Monitoring Center, Nanning, Guangxi
530025, China 5 Life Sciences Institute, Guangxi Medical University, Nanning, Guangxi 530021, China
6 Center for Genomic and Personalized Medicine, Guangxi Medical University, Nanning, Guangxi 530021, China
ABSTRACT Previous studies have identified multiple viruses in dead or severely diseased pangolins, but descriptions of the virome in healthy pangolins are lacking. This poses a greater risk of cross-species transmission due to poor preventive awareness and frequent interactions with breeders. In this study, we investigated the viral composition of 34 pangolins with no signs of disease at the time of sampling and characterized a large number of arthropodassociated viruses belonging to 11 families and vertebrate viruses belonging to eight families,including those with pathogenic potential in humans and animals. Several important vertebrate viruses were identified in the pangolins, including parvovirus,pestivirus, and picobirnavirus. The picobirnavirus was clustered with human and grey teal picobirnaviruses. Viruses with cross-species transmission ability were also identified, including circovirus, rotavirus, and astrovirus. Our study revealed that pangolins are frequently exposed to arthropod-associated viruses in the wild and can carry many vertebrate viruses under natural conditions. This study provides important insights into the virome of pangolins, underscoring the importance of monitoring potential pathogens in healthy pangolins to prevent outbreaks of infectious diseases in domesticated animals and humans.
Keywords: Pangolin; Virome; Arthropodassociated viruses; Cross-species transmission;Zoonoses
INTRODUCTION
Pangolins are currently the most captured and traded animals in the world and have been shown to carry a unique diversity of viruses, including SARS-CoV-2-related coronaviruses (Lam et al., 2020; Xiao et al., 2020). With ongoing tracing of the origin of SARS-CoV-2, attention has focused on pangolins.Pangolins are highly activity and consume a wide variety of insects, with a single adult consuming up to 70 million insects per year. Thus, pangolins are exposed to a huge variety of insect-associated viruses. Furthermore, adult pangolins can range over great areas (more than 10 km2), providing ample opportunities to interact with other wild animals, such as bats(Hassanin et al., 2021; Heath & Coulson, 1997), and to be exposed to unexpected viruses. Next-generation sequencing can be applied to reveal the entire virome in a sample in an unbiased manner (Harvey & Holmes, 2022; Zhang et al.,2018). Many viruses have been identified in dead or severely diseased pangolins, including coronaviruses (Lam et al., 2020;Xiao et al., 2020), human respiratory syncytial virus (Que et al., 2022; Ye et al., 2022), parainfluenza virus 5 (Wang et al., 2019), Sendai virus (Liu et al., 2019), canine parvovirus-2 (Wang et al., 2020), and pestiviruses (Gao et al., 2020; He et al., 2022). Given the restrictions on conducting experiments on this protected animal, clarifying the consequences of viruses in pangolins is difficult. However, for surveillance,conservation, and public health, viruses carried by healthy pangolins are of great significance as they can infect humans(Cdc, 2021; Islam et al., 2021). Early detection and identification of pathogens with potential cross-species transmission capabilities is the first step in preparing for future emerging infectious diseases. Understanding the virosphere and evolution of viruses is also essential. In this study, we investigated the diversity of viruses that may coexist in healthy pangolins but may also threaten humans and other animals.
MATERIALS AND METHODS
Sample collection
Pangolins smuggled to China from Southeast Asia were intercepted by Chinese customs in 2019. These pangolins were transported to the Guangxi Zhuang Autonomous Region Land Wildlife Medical Rescue and Epidemic Surveillance Research Center and were rescued under ethical approval(Wild Animal Treatment Regulation No. [2011] 85). Sample collection followed the guidelines listed in the Pangolins Rescue Procedure (November 2016). Throat swab, anal swab, and blood serum samples were obtained from the living pangolins. All samples were stored on dry ice before transfer to a -80 °C freezer for storage.
RNA extraction, library preparation, and sequencing
Each sample was subjected to RNA extraction using an AxyPrepTMMultisource Total RNA Miniprep Kit (Axygen,China) according to the manufacturer’s instructions and immediately transferred to a -80 °C freezer for storage. A sequencing library was constructed using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (USA). The library was subsequently sequenced on the Illumina NovaSeq 6000 (PE150) sequencing platform (USA).
Virus discovery and confirmation
For each library, adaptor sequences and low-quality bases were removed from the raw sequencing reads using Fastp(v0.20.0) (Chen et al., 2018). The remaining reads were compared against the SILVA database (www.arb-silva.de,v138.1) and pangolin genomes (GCF_014570555.1_YNU_ManPten_2.0 and GCF_014570535.1_YNU_ManJav_2.0) using Bowtie2 (v2.2.5) (Langmead & Salzberg, 2012) to remove reads associated with ribosomal RNA and host genomes, respectively. Unmapped reads werede novoassembled using MEGAHIT (v1.2.9) (Li et al., 2015, 2016).The assembled contigs were compared against the NCBI nonredundant protein database (nr) using Diamond BLASTx(v0.9.7) (Buchfink et al., 2015) to identify virus-associated contigs based on the top BLAST hits of each contig. A readmapping approach was used to estimate virus abundance.The reads remaining after ribosomal RNA and host genomes were removed were mapped to the assembled viral contigs,and virus abundance was quantified as the number of mapped reads per million total reads in the library (RPM).
Phylogenetic analysis
Based on the sequences obtained, nucleocapsid proteins,replication-associated proteins, and capsid proteins were used for phylogenetic analysis. Viral amino acid sequences were aligned with their respective virus families or genera based on the Diamond BLASTX results using MAFFT (v7.475) (Katoh et al., 2002). Phylogenetic trees were then estimated for the sequence alignment of each family or genus using maximumlikelihood in PhyML (v3.1) (Guindon et al., 2010), employing the LG model of amino acid substitution and subtree pruning and regrafting (SPR) branch swapping algorithm.
RESULTS
Viral metagenomics overview
The rescued pangolins, which exhibited no signs of disease at the time of sampling, were screened for pathogens. Samples were taken from three different batches of pangolins and collected within a day of their arrival at the rescue center. Due to the experience of different sampling personnel, the sample types varied from batch to batch. In total, 11 anal swabs(A1-A11), 11 throat swabs (T1-T11), and 12 blood serum samples (B1-B12) were collected from 34 pangolins and used for next-generation sequencing (Supplementary Table S1). In total, 1 747.1 Gb of data were obtained, and 52 virus families were identified. As plant-hosted viruses and bacteriophages are common in metagenomic sequencing and rarely pose biosafety risks, they were not further analyzed. The remaining 19 families were associated with insect or mammalian viruses(Figure 1A).
We investigated viral abundance in different tissues(Figure 1B). Results showed that similar proportions of vertebrate- and arthropod-associated viruses were found in the anal swabs. Members of theFlaviviridaeandParvoviridaefamilies accounted for the majority of vertebrate- and arthropod-associated viruses, respectively. Over 90% of viruses in the serum samples were vertebrate-associated, of whichCircoviridaeaccounted for more than 60%. Nine vertebrate-associated virus families accounted for 7.2% of total viruses in the blood, withRhabdoviridaefound to be dominant. Approximately 80% of the throat-swab viruses were vertebrate-associated, dominated byFlaviviridae, whileParvoviridaeaccounted for almost all the remaining 20% of arthropod-associated viruses.
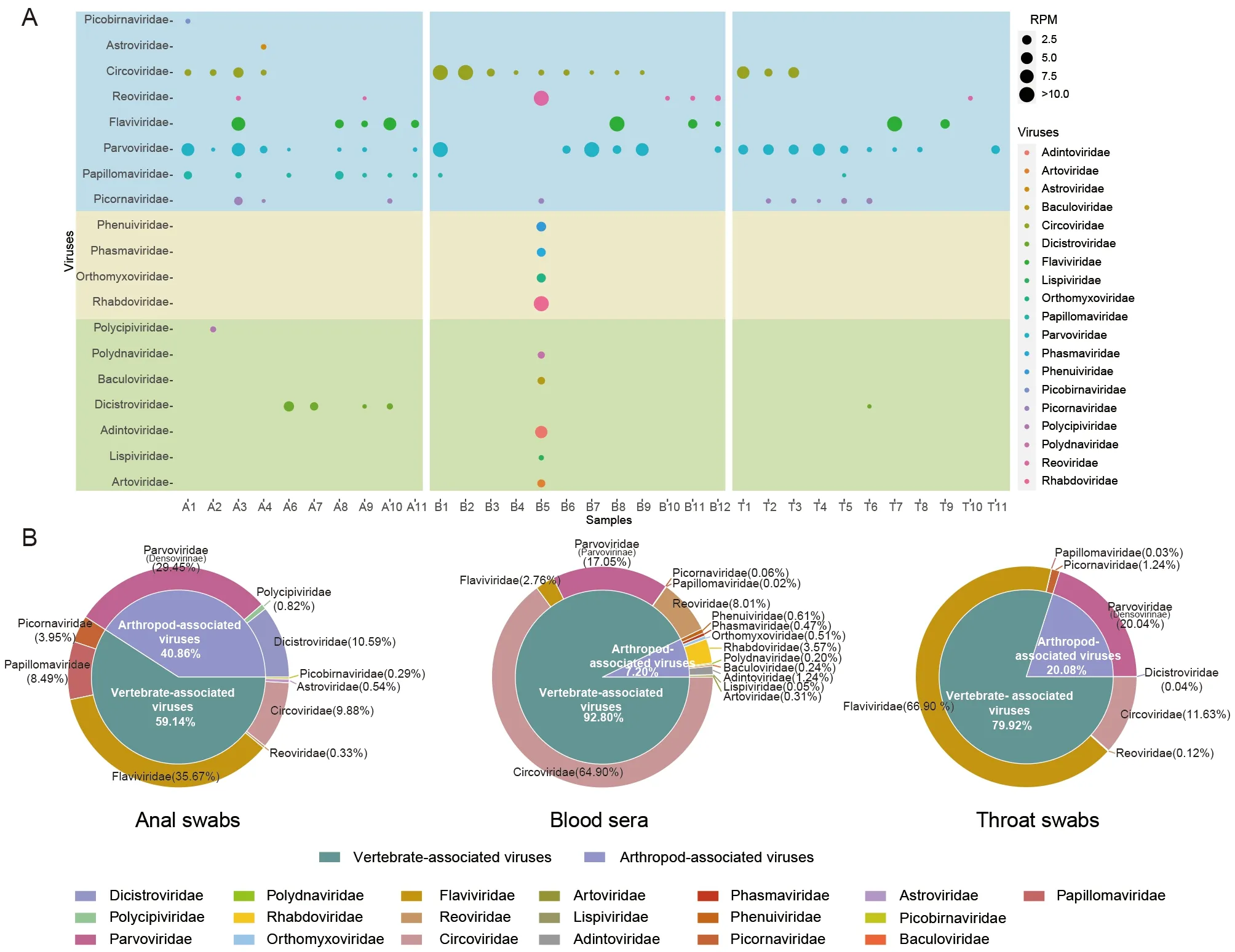
Figure 1 Distribution and abundance of viruses in pangolins
Diversity of insect viruses
We identified various viral sequences belonging to families of viruses mainly hosted by insects, includingPolycipiviridae,Polydnaviridae,Baculoviridae,Dicistroviridae,Adintoviridae,Lispiviridae, andArtoviridae(Figure 1A). For example,Polycipiviridaeis a family of non-segmented picorna-like viruses, primarily derived from arthropods, especially ants(Olendraite et al., 2019);Dicistroviridaeis a family of small non-enveloped viruses, whose natural hosts are invertebrates(Valles et al., 2017); andBaculoviridaeandPolydnaviridaeare hosted byLepidoptera,Hymenoptera, andDipteralarvae(Harrison et al., 2018; Stoltz et al., 1984).
Members of the familiesRhabdoviridae,Orthomyxoviridae,Phasmaviridae, andPhenuiviridaehave diverse hosts,including plants, arthropods, and vertebrates (Walker et al.,2018). Several viruses in these families were identified in this study, with a small genetic distance from arthropod-associated viruses (Figure 2).
Pangolin-specific viruses in families Parvoviridae,Flaviviridae, and Picobirnaviridae
Many of the novel vertebrate-associated viruses identified in the pangolins differed markedly from known viral genomes,representing a distinct branch of viruses in wild pangolins.
Viruses belonging to theParvoviridaefamily were identified in 23 samples and were distributed in theDensovirinaeandParvovirinaesubfamilies, which can be distinguished by their host associations, i.e., vertebrates (including humans) versus invertebrates (Cotmore et al., 2019).Densovirinaeviruses,which infect invertebrate hosts, were only identified in anal and throat swabs, whileParvovirinaeviruses, which infect vertebrate hosts, were only identified in six serum samples.Four novel near-completeDensovirinaegenomes and two novel near-completeParvovirinaegenomes were obtained,which clustered on a new branch with theAmbidensovirusandCopiparvovirusgenera, respectively (Figure 3).Densovirinaemay have been sourced from pangolin food, whileParvovirinaemay be naturally circulating in pangolin populations.

Figure 2 Interspecific phylogenetic relationships of four virus families associated with arthropod hosts
Pestiviruses, which belong to the familyFlaviviridae, infect pigs, ruminants, and even-toed ungulates (Schweizer &Peterhans, 2014), and appear to be widespread in pangolins,with a high degree of pathogenicity and potential for lethality(Gao et al., 2020; He et al., 2022). In this study, we detected a diverse group of novel pestiviruses in the different pangolin samples. Using the completePestivirusgenome obtained from sample T7 as a reference sequence, we found thatPestivirusgenome segments from different samples shared 65%-100% nucleotide identity with the T7 reference(Figure 4A). Conserved amino acid sequences in the RNAdependent RNA polymerase (RdRP) in theFlaviviridaeand pangolinPestivirusgenomes were used for evolutionary analysis (Simmonds et al., 2017). Results showed that the pangolinPestivirusidentified in this study was clustered in a sister clade with the pathogenic Dongyang pangolinPestivirus(DYPV) (Figure 4B), suggesting that different and specificPestivirusgroups circulate in wild pangolin populations with different degrees of virulence.

Figure 3 Phylogenetic relationships of viruses in the family Parvoviridae

Figure 4 Pestivirus-associated viruses detected in pangolin metagenomics data and phylogeny of the family Flaviviridae
Picobirnavirus, the only recognized genus in the familyPicobirnaviridae, is widely distributed in humans and mammals. Here, we detectedPicobirnavirusin one pangolin anal swab, which showed the closest genetic distance to grey teal and humanPicobirnavirusin the capsid-based phylogenetic tree (Figure 5).Picobirnavirusis routinely detected in stool samples (Navarro et al., 2017); however,there is growing evidence that it is also present in the upper respiratory tract (Berg et al., 2021; Huaman et al., 2021).Picobirnavirusspecies are considered opportunistic due to their association with diarrhea in humans and animals;however, their epidemiology and pathogenicity have not yet been characterized (Kashnikov et al., 2020). Therefore, this rapidly evolving and easily transmitted virus requires continued attention to prevent the emergence of diseasecausing variants. Our evidence showed thatPicobirnavirusis also naturally present in pangolins.
Cross-species transmission of viruses in pangolins
Circoviruswas detected in all three sample types. Nine completeCircovirussequences were obtained, which showed high similarity (96.19%-98.61%) to porcine circovirus 1(PCV1, NC_001792) and a close genetic relationship with the PCV1 group inCircovirusbased on phylogenetic analysis of Rep and Cap protein sequences (Figure 6A, B). PCV1 is apathogenic in pigs, and broadly distributed in ruminants,rodents, canines, insects, and other species (Zhai et al.,2019). Our study demonstrated that PCV1 is also highly transmissible in pangolins and may be present due to pigassociated transmission and subsequent spread in pangolin populations.
A partial astrovirus genome was identified from an anal swab, which contained 105 sequence reads matching theAilurus fulgensastrovirus genome (GenBank accession No.MZ357116), covering all three open reading frames (ORFs)(ORF1a, RdRp, and capsid proteins) (Figure 7A). The partial astrovirus genome of the pangolin shared approximately 96%nucleotide identity withAilurus fulgensastrovirus 1, previously identified in a panda-breeding location (Zhao et al., 2022).Based on phylogenetic analysis of the capsid gene(Figure 7B), the pangolin astrovirus clustered in
Mamastrovirus group IIwith theAilurus fulgens, feline, and raccoon dog astroviruses. The presence of similar astroviruses in different species indicates cross-species transmission potential in multiple animals. Notably, similar astroviruses have been identified from different sources,suggesting possible complex transmission pathways in the natural environment.
Genome segments of rotaviruses, which only infect vertebrates, were identified in six samples (Figure 8A) and shared 77%-91% nucleotide identity with known rotaviruses from different hosts (e.g., bat, human, horse, sugar glider,dog, and pig). The rotavirus genome contains 11 discrete segments of linear double-stranded RNA (dsRNA), and genomic reassortment and rearrangement between segments can enhance its evolutionary drive and potential for crossspecies dispersal (Ghosh & Kobayashi, 2014). The identification of rotavirus in pangolins suggests their potential as incubators for rotavirus-producing fragment exchange. In addition, several new members of theReoviridaefamily were identified with a close genetic distance to plant- and arthropod-hosted viruses (Figure 8B).

Figure 5 Phylogenetic analysis of Picobirnavirus
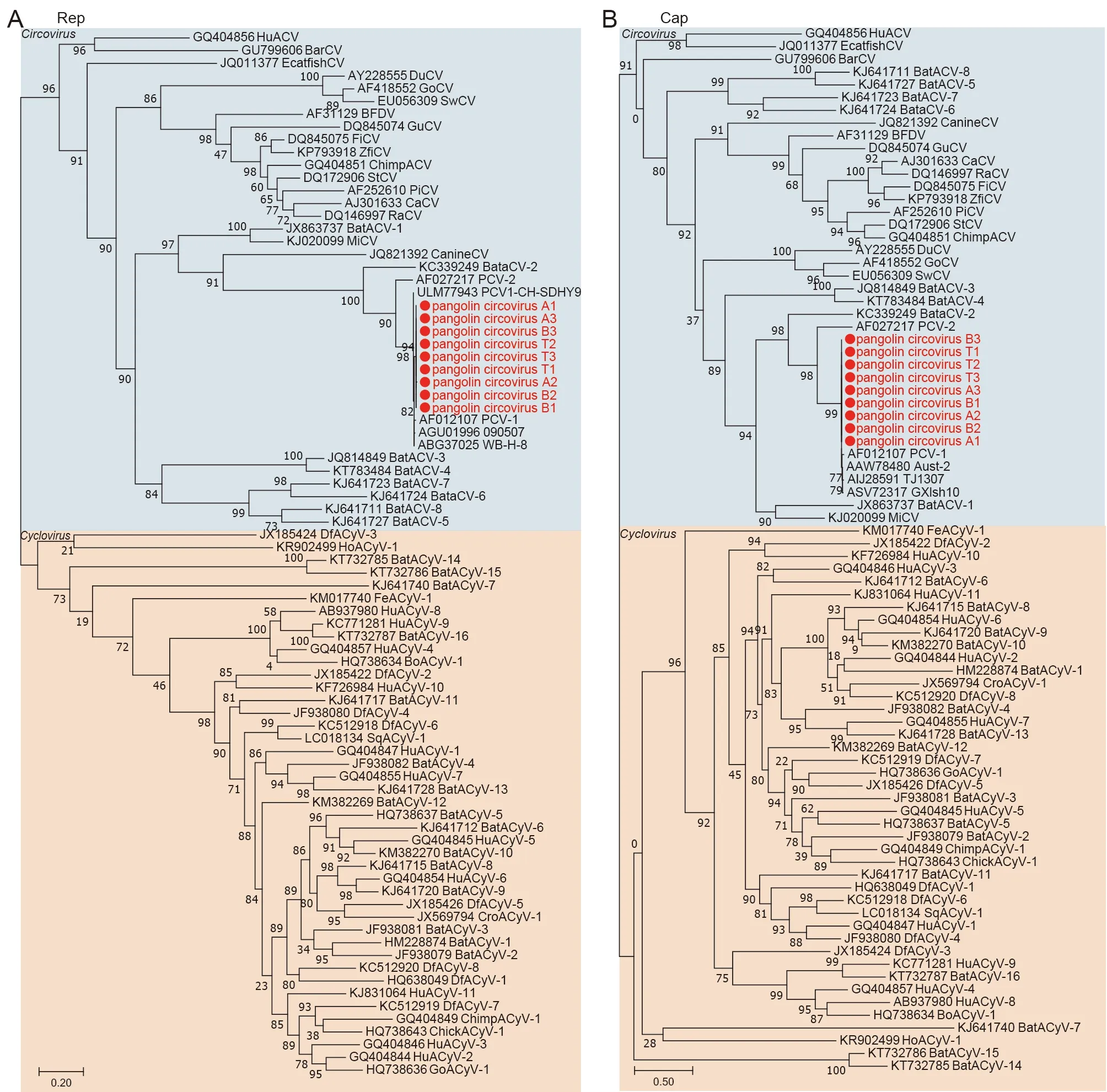
Figure 6 Phylogenetic analyses of Circovirus

Figure 7 Analysis of astrovirus-associated viruses

Figure 8 Abundances of viruses from Reoviridae family in pangolins
DISCUSSION
Disease outbreaks associated with wildlife pose a significant burden on global health and security. Early detection and identification of pathogens with potential cross-species transmission capabilities is the first step in preparing for future infectious diseases and is also important for understanding the virosphere and evolution of viruses. In the present study, we explored the viromes of 34 pangolins with no apparent disease and identified several significant arthropod- and vertebrate-associated viruses.
Previous virome investigations of diseased pangolins have focused on vertebrate-hosted viruses, includingReoviridae,Parvoviridae,Papillomaviridae,Circoviridae,Flaviviridae, andPicornaviridae(Liu et al., 2019; Ning et al., 2022; Yang et al.,2021). In this study, we provided more detailed information and confirmed that these viruses are carried by healthy pangolins. In contrast to our findings, previous research has indicated that DYPV is associated with hemorrhagic disease(Gao et al., 2020). A possible explanation for this discrepancy is that pestiviruses are opportunistic pathogens or different pestivirus genomes show different virulence to pangolins. In this study, members of thePicobirnaviridaeandAstroviridaefamilies were identified in pangolins for the first time. These two families have a wide host recognition range. The pangolin picobirnavirus clustered with the sister clade of the human picobirnavirus, and the pangolin astrovirus showed 96% nucleotide identity with theAilurus fulgensastrovirus,suggesting a high risk of cross-species transmission.
In addition, many novel arthropod-associated viruses were identified in the pangolins. Notably, the serum samples showed an unexpected abundance of arthropod-associated viruses, possibly from insect bites (most likely mosquito or tick) (Hassan et al., 2013). The immune function of pangolins is defective (Choo et al., 2016; Haley, 2022), which may contribute to the viral storage and compatibility of pangolin blood and provide a new transmission pathway for the spread of viruses. Unfortunately, metagenomics does not provide direct evidence of host infection and it is difficult to conduct further experiments to confirm the occurrence of possible infections. However, our study has demonstrated, to least to some extent, that these viruses exist in healthy pangolins, and viral evolution and cross-species transmission may occur.
Although pangolin susceptibility to coronaviruses has been demonstrated many times, the positivity rates vary among populations, ranging from 25% to 81% in different batches of dead pangolins (e.g., 17/21 (Xiao et al., 2020), 5/18 (Lam et al., 2020), 3/12 (Lam et al., 2020), and 7/15 (Nga et al.,2022)). Two large-scale surveys of pangolins found that only a few individuals tested positive for coronavirus among hundreds of pangolin samples (e.g., 1/161 (Shi et al., 2022)and 4/163 (Peng et al., 2021)). In the current study, we did not detect the presence of coronavirus in the pangolin samples.The reasons for differences in the coronavirus positivity rates among tested pangolin populations are likely complex, possibly impacted by (i) the widespread distribution of pangolin coronaviruses in specific geographic environments,and (ii) the overcrowding of this normally solitary animal during smuggling, thus facilitating the spread of viruses. As smuggled pangolins cannot be traced back to their origin, it is very difficult to know exactly how pangolins are infected with coronavirus. Whether human activities amplify the risk of viruses of natural origin requires further study.
Our results indicate that pangolins with no signs of disease can still carry many viruses. We investigated the viromes of pangolins and possibility of infectious disease outbreaks. This study not only provides details regarding the monitoring of pangolin health, but also reveals the possible health risks to other animals and humans that may encounter pangolins.
DATA AVAILABILITY
The data that support our findings were deposited in the NCBI database under BioProjectID PRJNA881017, as well as the Genome Sequence Archive (GSA) under accession no.PRJCA010128, and the Science Data Bank under DOI:10.57760/sciencedb.j00139.00041. The consensus sequences in this study were deposited in GenBank under accession numbers OP474153-OP474178 and in the NGDC Genome Warehouse (GWH) (https://ngdc.cncb.ac.cn/gwh/)under accession numbers GWHBJSI01000000-GWH BJTI01000000 (Supplementary Table S2).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Y.G.T., M.S., T.C.Q., and Y.L.H. designed and supervised the research. T.C.Q. collected the samples. W.L.L., Y.J.L., Y.J.H.,Q.H.T., Y.L., and Y.B. processed the samples. F.J.T. and J.L.performed data analysis, genome assembly, and phylogenetic analysis and contributed to data interpretation and wrote the paper. Y.G.T., M.S., T.C.Q., and Y.L.H. edited the paper. All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Optimization of sgRNA expression strategy to generate multiplex gene-edited pigs
- Antiviral effects of natural small molecules on aquatic rhabdovirus by interfering with early viral replication
- Chronic lithium treatment ameliorates ketamineinduced mania-like behavior via the PI3K-AKT signaling pathway
- A glimpse into the biodiversity of insects in Yunnan: An updated and annotated checklist of butterflies(Lepidoptera, Papilionoidea)
- Unveiling the functional and evolutionary landscape of RNA editing in chicken using genomics and transcriptomics
- Animal models of Alzheimer’s disease: Applications,evaluation, and perspectives