CRISPR/Cas9基因编辑工具开发与应用研究进展
2022-11-24庞紫燕官阳阳孙嘉磊王琰
庞紫燕,官阳阳,孙嘉磊,王琰
(武汉大学 生命科学学院 细胞稳态湖北省重点实验室,湖北武汉 430072)
规律间隔成簇短回文重复序列(Clustered Regularly Interspaced Short Palindromic Repeats,CRISPR)及其相关蛋白(CRISPR-associated protein,Cas)是原核生物抵御病毒感染或噬菌体入侵产生的一种适应性免疫系统,其主要通过适应、表达、干扰3个基本阶段来保护细菌免受病毒反复攻击[1]。自被发现以来,研究人员不断对CRISPR/Cas系统进行优化升级,使其成为了分子生物学领域重要的基因编辑工具之一,其主要通过特异位点识别、靶标基因切割和修复3个步骤实现基因编辑[2]。
由于Ⅱ型CRISPR/Cas9基因编辑系统组成相对简单,操作简便,编辑效率高,在基因编辑中得到了广泛的研究和应用[1]。近年来,研究人员不断发掘出更多种类的Cas9蛋白,并对其进行优化改造,以扩大识别基因组的范围,实现精准高效的基因编辑[2]。此外,一系列基于CRISPR/Cas9系统的基因编辑工具也被不断开发出来,如能对单个碱基进行转变的碱基编辑器(Base Editors,BEs);在不造成基因组断裂的情况下实现特定靶点碱基替换、片段插入或删除的Prime编辑器(Prime Editors,PEs)等,适用于体内、体外各种基因编辑活动,并可用于疾病的治疗,具有巨大的应用潜力[3-5]。本文将对CRISPR/Cas9系统的组成、结构、基因编辑机制和不同Cas9蛋白及变体进行概述,并着重介绍基于CRISPR/Cas9系统开发的基因编辑工具和相关应用。
1 CRISPR/Cas9系统基因编辑机制
CRISPR/Cas9系统主要由引导RNA(guide RNA,gRNA)和Cas9蛋白组成。Cas9蛋白本质上是一种多域DNA核酸内切酶,负责切割目的基因形成双链断裂,也被称为遗传剪刀[6]。在使用CRISPR/Cas9系统进行基因编辑时,研究人员把crRNA和反式激活crRNA(transactivating crRNA,tracrRNA)组合起来设计成单个引导RNA(single guide RNA,sgRNA),用来引导Cas9蛋白对目的基因进行切割[6]。
CRISPR/Cas9系统主要通过识别、切割和修复3个步骤进行基因编辑[2]。sgRNA通过其5’-crRNA与目标基因碱基互补配对来识别靶序列,引导Cas9核酸酶定位到靶序列。如图1所示,在RNA-DNA杂交体形成后,Cas9蛋白在PAM序列上游的3 bp处对DNA进行切割,产生一个平末端双链断裂(Double Strand Break,DSB)[7],随后目标基因通过非同源末端连接(Non-Homologous End Joining,NHEJ)途径或同源定向修复(Homology-Directed Repair,HDR)途径进行修复。在没有修复模板的情况下,双链断裂通过NHEJ途径重新连接,它是细胞内的主要修复机制,在各个细胞周期都很活跃,但可能导致连接位点的随机插入缺失突变,导致移码突变或无义突变,可用于基因敲除[8]。HDR通常仅在分裂细胞中活跃,发生频率较低,但可以利用外源修复性模板在目标基因上进行精确的插入或替换[9]。
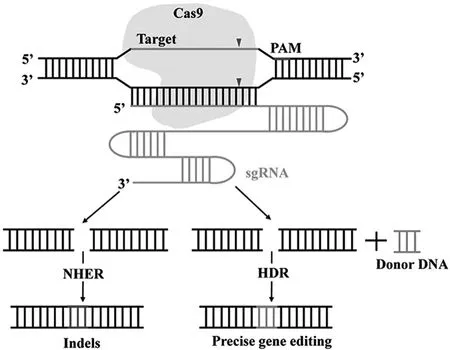
图1 CRISPR/Cas9基因编辑原理示意图
化脓性链球菌Cas9(Streptococcus pyogenesCas9,SpCas9)是第一个用于基因编辑的Cas9蛋白,研究人员对其进行突变改造,以提高靶向特异性、降低脱靶效应、识别不同PAM序列,生成了一系列新的变体,如SuperFi-Cas9、xCas9等[6]。此外,研究人员还设计了具有特殊功能Cas9蛋白变体[10],如核酸酶活性丧失的Cas9(nuclease dead Cas9,dCas9)可以与其他分子融合,使其在特定位点发挥作用;只能对目的基因进行单链切割的切口酶Cas9(Cas9 nikase,nCas9)在基因编辑过程中可激活细胞内高保真的HDR途径进行修复,所以常用于高精准的基因编辑[11]。如北京大学胡家志课题组将SpCas9与核酸外切酶TREX2融合生成核酸内外切酶Cas9TX变体,其几乎可以消除染色体易位和大片段缺失的发生,大大提高了基因编辑的安全性[12]。
2 基于CRISPR/Cas9的基因编辑工具开发
2.1 转录调控
转录调节因子可以和dCas9融合形成复合物,在gRNA引导下,靶向目的基因的启动子或增强子序列,以沉默(CRISPRi)或激活(CRISPRa)特定目标基因的表达,从而进行体内或体外的筛选研究[13]。CRISPRa系统通过转录激活因子(如VP16、VPR和p300等)来促进转录翻译[14]。相反,CRISPRi系统多与 Krüppel相关盒(Krüppel-Associated Box,KRAB)融合使用,诱导组蛋白甲基化和去乙酰化,从而抑制RNA聚合酶与增强子或启动子区域的结合,使转录失活[15]。近期某研究小组使用CRISPRi筛选发现含溴结构域蛋白2(Bromodomain-Containing Protein 2,BRD2)是SARS-CoV-2感染宿主的重要调节因子,为治疗COVID-19的感染提供了又一重要靶点[16]。此外,将荧光蛋白等标记蛋白与dCas9酶融合,可以实现染色质活细胞成像和亚细胞定位[17]。
2.2 转座与重组
CRISPR/Cas9系统也可实现较长DNA片段的靶向插入。由转座酶与dCas9融合开发的Cas-转座子系统可以将外源DNA片段插入到特定位点,如marer家族转座酶Himar 1与dCas9重组后,可以在大肠杆菌和哺乳动物细胞中将所需片段特异地插入到目标TA基序中[18]。如斯坦福大学丛乐教授课题组根据噬菌体能够精确的向宿主细胞重组的现象推断出微生物重组的关键酶——单链退火蛋白(the Single-Strand Annealing Protein,SSAP)可用于哺乳动物细胞中的无切割基因编辑,将其与dSpCas9系统相结合,开发出新型dCas9-SSAP基因编辑工具。如图2所示,该工具由SSAP与MS2噬菌体外壳蛋白(MS2 Coat Protein,MCP)形成的二元复合物,dCas9蛋白以及含有MS2茎环序列的gRNA三部分组成,MS2序列可以招募多个SSAP-MCP复合物,从而允许多个SSAP与dCas9-gRNA形成复合物,随后SSAP附着到供体DNA上,在无任何DNA断裂的情况下进行DNA重组交换,最终实现长达1 kb左右的长序列精准编辑。而选用较小的dSaCas9,并对SSAP蛋白进行截断突变,开发出了更易递送的dSaCas9-mSSAP系统,可以最大限度地减少体内基因编辑治疗疾病中的意外突变[19]。
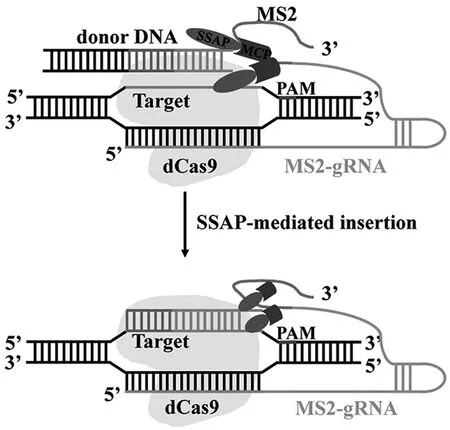
图2 dCas9-SSAP的示意图模型
2.3 碱基编辑
基于CRISPR/Cas9系统开发的BEs无需供体DNA模板即可实现单个碱基的替换,并且在编辑过程中不产生DSB,大大降低了indels的发生,可用于单基因突变疾病的治疗[4]。目前已经开发了胞嘧啶碱基编辑器(Cytosine Base Editors,CBEs)、腺嘌呤碱基编辑器(Adenine Base Editors,ABEs)和糖基化酶碱基编辑器(Glycosylase Base Editors,GBE)3种工具,如图3所示[3]。
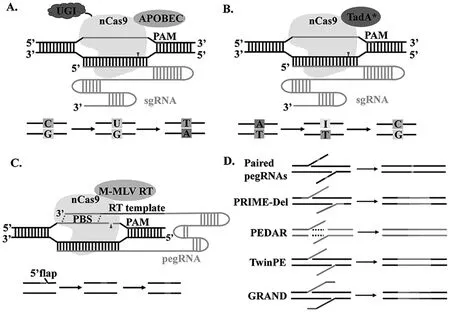
图3 基于CRISPR/Cas9的基因编辑工具示意图
CBEs由dCas9或nCas9与大鼠来源的胞嘧啶脱氨酶——载脂蛋白B mRNA编辑酶催化亚基1(Apolipoprotein B mRNA Editing Enzyme Catalytic Subunit 1,APOBEC1)以及尿嘧啶糖基化酶抑制子(Uracil DNA Glycosylase Inhibitor,UGI)融合组成。如图3A所示,在dCas9或nCas9结合特定序列后,胞嘧啶脱氨酶首先将C脱氨基生成U,接着在DNA复制过程中再变为T,最后将原来反义链中的G转变为A,最终实现由C-G到T-A的精准碱基替换。其中,UGI主要防止DNA复制过程中U的切除,增加碱基替换效率[20]。自初代CBE问世以来,研究人员已经通过更换不同Cas蛋白、使用不同来源的脱氨酶、对脱氨酶进行突变改造、优化两者之间的连接子以及增加UGI数量等方式开发出了不同的版本的CBE,以增加编辑效率,拓宽脱氨酶催化活性窗口,减少脱靶效应和旁观效应,如SECURE(R33A/K34A)、A3A(Y130F)-BE3 以及 FERNY-BE4max[21-23]。
ABEs由nCas9与腺嘌呤脱氨酶融合构成,如图3B所示,在sgRNA的引导下,融合蛋白上的腺嘌呤脱氨酶可将靶DNA上的A脱氨基变成I,而在DNA复制过程中I通常被当做G,最后完成A-T碱基对到G-C碱基对的准确替换。由于目前没有发现直接作用于DNA的腺嘌呤脱氨酶,ABEs的开发难度更大。初代ABE1.2的腺嘌呤脱氨酶TadA*是在大肠杆菌的tRNA腺苷酸脱氨酶(ecTadA)上进行突变改造而来的,经过7轮升级改造开发出了ABE7.10,其由野生型TadA和TadA*的异二聚体与nCas9融合组成(TadA-TadA*-nCas9),碱基编辑效率和特异性大大提高[24]。此外,研究人员尝试使用能识别不同PAM序列的Cas9蛋白及变体以扩大ABEs在人类基因组中的编辑位点,如NG-ABE、xCas9-ABE与ScCas9-ABE,但由于ABE与这些变体Cas9的兼容性有限,其编辑效率明显降低[25-26]。近期开发的新版本ABE8e与ABE8s使用具有更大脱氧腺苷脱氨活性的TadA变体,与其他Cas9的兼容性较好,目前已经衍生出了多个版本的ABE8e,与ABE7.10相比,碱基编辑效率进一步提高,且具有更广泛的活性窗口[27-28]。然而,ABE8e的脱靶效应随着脱氧腺苷酶活性的提高而增加,因此研究人员在TadA上引入V106W点突变来改善脱靶编辑[28]。
最近,研究人员还开发出了3种基于CBEs和ABEs的双碱基编辑器——SPACE、A&C-BEmax和Target-ACEmax,它们可以在同一等位基因中同时诱导C-T和A-G的碱基转变,且都由胞嘧啶脱氨酶、腺嘌呤脱氨酶、nCas9与UGI 4部分组成,可以实现较为复杂的基因编辑[29-31]。
GBE由nCas9、胞嘧啶脱氨酶和尿嘧啶-DNA-糖基化酶(Uracil-DNA Glycosylase,Ung)组成,原理类似于CBEs,即先通过胞嘧啶脱氨酶将C转变为U,随后Ung再将U碱基切除,形成无嘌呤/无嘧啶位点,进而激活DNA损伤修复机制,完成碱基转变[32-33]。研究发现,使用活化诱导的胞苷脱氨酶(Activation-Induced cytidine Deaminase,AID)构建的AID-nCas9-Ung系统可在大肠杆菌中将C颠换为A,通过改造构建出可以在哺乳动物细胞中实现C到G碱基颠换的APOBEC-nCas9-Ung系统[32]。另外,有研究发现使用rAPOBEC1(R33A)突变体构建的CGBE1系统也可实现C-G碱基颠换,并且可以降低脱靶效应;去除Ung的miniCGBE1系统仍保留C-G碱基颠换活性,并且能有效降低indel的风险[33]。
2.4 Prime编辑器
总的来说,碱基编辑器替换碱基的种类有限,为了克服这一缺点,刘如谦教授团队开发出了PEs,其同样在不产生DSB的情况下能实现短片段的插入删除和所有可能的碱基转变,大大扩展了基因组编辑范围。如图3C所示,PEs由工程逆转录酶、nCas9和pegRNA(prime editing guide RNA)3部分构成,其中pegRNA是在sgRNA的3′末端增加一段与靶DNA链3’末端互补的引物结合位点(Primer Binding Site,PBS)以及一段携带有目标突变的序列,既可起始逆转录过程,又能作为逆转录的模板。PEs编辑原理为在pegRNA的引导下,nCas9定位到靶DNA链以形成一个切口,切口的3’端与引物结合位点互补使逆转录酶将模板信息复制到DNA链上,随后细胞内结构特异性核酸内切酶(如FEN1)或5’核酸外切酶(如EXO1)去除切口处剩下的5’尾巴,DNA连接酶将3’尾巴整合到DNA双链上,最后细胞内DNA修复机制启动,最终实现精准且稳定的基因编辑[34-35]。
PE1由nCas9与野生型莫洛尼鼠白血病病毒逆转录酶(Moloney Murine Leukaemia Virus Reverse Transcriptase,M-MLV RT)融合而成,编辑能力较低,通过5个突变(D200N/L603W/T330P/T306K/W313F)改造开发出了编辑效率更高的PE2。PE3则是在PE2的基础上引入了靶向非编辑链的sgRNA,使nCas9同时在非编辑链上产生切口,加快触发DNA修复机制,从而进一步提高编辑效率。为了降低基因编辑中indels发生的概率,研究人员将PE3中靶向非编辑链的sgRNA替换为靶向经pegRNA引导编辑后产生的新DNA链的sgRNA,进而开发出了PE3b[35]。
由于pegRNA的3’暴露在细胞中,容易被核酸外切酶降解,刘如谦教授团队使用一个连接接头将结构化的RNA基序(evopreQ1或mpknot)连接到pegRNA的3’末端,以增强其稳定性,编辑效率可提高1.5~2.0倍[36]。此外,DNA错配修复系统相关基因强烈地抑制PEs的编辑效率,并促进indels的发生,通过瞬时表达显性阴性突变型的错配修复蛋白MLH1(MLH1dn)开发的PE4(PE2+MLH1dn)和PE5(PE3+MLH1dn)两个编辑系统,基因编辑效率显著提高,且降低了indels的出现。而通过改变RT密码子使用、SpCas9突变、核定位序列、nCas9和逆转录酶之间连接肽的长度和组成以及epegRNA优化得到的PEmax架构,可将编辑效率进一步提高2倍[37]。此外,高彩霞团队发现在植物中使用一对分别靶向目的基因两条DNA链的pegRNA可以显著提高PEs编辑效率,并且实现短片段的插入[38]。
人类遗传变异还包括基因的插入缺失、重复及倒位等多涉及大片段DNA的变异,治疗此类遗传疾病需要开发出能够精确地删除插入或复制的DNA序列,并修复被破坏基因位点的基因编辑工具。(1)PRIME-Del系统。其在PE2基础上使用一对分别靶向目的基因两条DNA链的pegRNA来诱导两个nCas9切口之间的基因删除,重要的是需保证pegRNA上逆转录的模板与另一条pegRNA靶向的切口位点序列同源,以促进DNA修复整合。实验结果显示该系统可以精准删除长达10 kb的DNA片段,同时可以在pegRNA逆转录模板上引入短片段的插入[39]。(2)由原始Cas9与逆转录酶的融合蛋白以及一对pegRNA组成的PEDAR系统。该系统要保证逆转录酶能够沿模板在Cas9的切口两侧各合成一段粘性末端,经过退火和DNA修复连接后,可以实现精确删除长片段的同时插入外源DNA。实验结果显示,PEDAR系统可删除长达10 kb的DNA片段,同时插入60 bp左右的外源DNA,但与PRIME-Del系统相比,它有较高的indels风险[40]。(3)TwinPE系统。该系统使用一对pegRNA靶向目的基因的两条链,与PRIME-Del不同的是,TwinPE的pegRNA逆转录模板不需与DNA链同源,因此TwinPE系统具有更灵活的模板序列,以及更大的长片段插入能力。受限于PE2系统插入片段长度的限度,TwinPE系统最多插入100 bp的DNA片段。为了能够插入更长的片段,研究人员将TwinPE系统与位点特异性整合酶Bxb1结合,使其能够整合大于5 kb的目的基因,并实现了长达40 kb的基因倒置[41]。(4)GRAND编辑系统。与TwinPE系统相似,其两条pegRNAs的逆转录模板仅部分互补结合,经DNA修复机制,完成20 bp到1 kb DNA的靶向插入[42]。
3 疾病治疗
CRISPR/Cas9在构建动物疾病模型,寻找疾病治疗的有效靶点,治疗许多遗传病、心血管疾病、传染病以及癌症中发挥重大作用[43]。
3.1 体外基因编辑治疗
镰刀型细胞贫血病(Sickle Cell Disease,SCD)是一种由编码β-珠蛋白的HBB基因上第一个外显子上发生单碱基突变引起的常染色体隐性遗传病,在低氧条件下,会出现贫血、疼痛、免疫缺陷、多器官衰竭甚至早亡的症状[44]。有研究利用CRISPR/Cas9系统在祖细胞和永生化人红细胞系中筛选出了可作为SCD治疗靶点的转录因子HRI和ZNF410[45-47]。此外,使用一种新的ABE8e-NRCH对患者来源的造血干细胞和祖细胞(Hematopoietic Stem and Progenitor Cells,HSPCs)进行单碱基编辑,把致病突变缬氨酸(GTG)转换为非致病突变丙氨酸(GCG),然后把编辑后的HSPCs移植到小鼠体内,其基因编辑效率达到68%,小鼠体内镰状红细胞明显减少,研究表明离体碱基编辑造血干细胞有望实现一次性永久治疗SCD[48]。
一项利用CRISPR/Cas9系统治疗β-地中海贫血(Transfusion-Dependent β-Thalassemia,TDT)和SCD的临床试验也取得了喜人的结果。在这项研究中,研究人员使用CRISPR/Cas9系统在从患者体内分离的HSPCs中对B细胞淋巴瘤11A(B-cell lymphoma 11A,BCL11A)基因进行编辑,以恢复患者胎儿血红蛋白的产生,抑制镰状血红蛋白聚合,随后把编辑后的HSPCs注射到一名TDT患者和一名SCD患者体内。经过治疗,两名患者体内维持高水平的胎儿血红蛋白表达,血管阻塞发作消除[49]。
在一项治疗晚期癌症患者的人体实验中,研究人员基于通过CRISPR/Cas9系统去除免疫检查点调节基因来增强细胞毒性T淋巴细胞的天然抗肿瘤反应的策略,对3名患者血液中提取的T细胞进行改造,删除了干扰对抗癌细胞的基因(TRAC、TRBC和PD-1),然后将改造后的T细胞重新注入患者体内,改造后的T细胞可以靶向特定抗原并杀死癌细胞,没有任何副作用,可存于体内长达9个月[50]。
3.2 体内基因编辑治疗
前蛋白转化酶枯草杆菌溶菌素9(Proprotein Convertase Subtilisin Kexin 9,PCSK9)是一种参与胆固醇代谢的基因,人体中自然发生的功能丧失的PCSK9突变没有明显的不良健康后果,且携带此类突变的人血液中的低密度脂蛋白胆固醇水平较低,动脉粥样硬化性心血管疾病的患病风险较低,这表明破坏PCSK9基因可能是治疗家族性高胆固醇血症的一种重要策略[51]。实验人员用脂质纳米颗粒将ABE mRNA与sgRNA有效递送到灵长类动物体内,并精确地在体内引入单核苷酸PCSK9功能丧失突变,从而降低了血清中PCSK9和低密度脂蛋白胆固醇的水平,并在8个月内保持稳定[52-53]。
伦敦大学的研究人员进行了首个体内利用CRISPR/Cas9系统对人类基因进行编辑以治疗ATTR淀粉样变性的临床试验。ATTR淀粉样变性是一种单基因疾病,其特征是错误折叠的转甲状腺素蛋白(Transthyretin,TTR)在人体神经系统、肾脏以及心脏等器官和组织中异常沉积,最终导致器官衰竭和死亡。该临床实验使用NTLA-2001来降低血清中TTR蛋白水平以治疗ATTR淀粉样变性。NTLA-2001由一个具有肝脏靶向递送的脂质纳米颗粒(lipid Nanoparticle,LNP)递送系统组成,其内包裹一个靶向人类TTR基因的sgRNA和一个Cas9蛋白的mRNA序列。LNP递送系统通过静脉注射到血液循环中,其表面与血浆中载脂蛋白E相结合,然后被肝脏表面的低密度脂蛋白受体摄取,随后LNP分解释放内容物,所以该系统可在体内靶向肝脏递送各种治疗性RNA,如siRNA和mRNA[54]。TTR几乎只在肝脏中表达,所以使用LNP这种以肝脏为靶点的递送系统可以最大限度提高疗效,同时减少全身毒性。6名患者低剂量注射NTLA-2001后血清TTR水平降低52%,高剂量注射后则降低了87%,仅有轻度不良反应[55]。
4 CRISPR/Cas9应用的挑战与展望
尽管CRISPR/Cas9技术在基因组编辑上前景广阔,但缺乏安全有效的靶向递送系统、脱靶效应、免疫原性以及癌症和伦理问题一直是将该技术扩展到临床应用的主要障碍[56]。
将CRISPR系统有效地传递到特定的细胞和器官以进行基因编辑是治疗过程中的一大挑战,理想的递送载体应该是安全的,具有高效的靶向性、高效率和生物可降解[57]。根据治疗方法的不同,递送方法可能会有很大差异。目前,将CRISPR/Cas9复合物递送到细胞中的方法主要有物理、化学和病毒载体3种。
非病毒(物理和化学)方法更适合离体基因编辑疗法[58],通常在患者体内分离的衍生细胞中进行基因编辑,扩增后再重新引入患者体内,可以使用质粒载体、核糖核蛋白复合物、纳米颗粒等将CRISPR系统递送到细胞中[59]。离体递送策略有几个优点:(1)递送发生在细胞水平上,效率更高,可以在临床应用之前快速评估;(2)CRISPR系统没有被引入体内,降低了对其他组织的非特异性基因编辑;(3)使用来自患者的细胞可以减少免疫反应的出现。然而,患者来源的细胞可能难以分离、培养和扩增,极大地限制了可治疗疾病的范围。此外,当这些基因组编辑的细胞被重新引入患者体内时,通常只有一小部分细胞能发挥功能[60]。
体内基因编辑可以应用于更广泛的疾病治疗,CRISPR系统主要由纳米颗粒或病毒载体传递,常使用病毒载体有腺病毒(Adenovirus,AD)、腺相关病毒(Adeno-Associated Virus,AAV)和慢病毒(Lentivirus,LV)载体,相比于物理和化学方法,其递送效率更高[61]。AAV是最常用的病毒载体,因为它的免疫原性更低且不整合到宿主细胞基因组中[62]。然而,AAV最多可包装4.7 kb的外源单链DNA,而Cas9蛋白较大,包装困难。为了解决这一限制,常将sgRNA和Cas9分别包装成两个单独的病毒,然后将它们共同注射到小鼠体内[63]。但是,最近有研究报道AAV基因组能够整合到Cas9产生的双链断裂位点[64],且体内有针对AAV衣壳蛋白的免疫反应的产生[65],说明病毒载体的安全性和有效性有待解决。刘如谦教授团队新开发了一种工程化的无DNA病毒样颗粒(engineered DNA-Free Virus-Like Particles,eVLPs),其可以在多种细胞类型和器官中实现高效的基因编辑,并最大限度地减少脱靶和DNA整合的风险,可用于治疗性核糖核蛋白的包装以及体外体内的递送,整合了病毒和非病毒载体的优势,具有广泛的应用前景[66]。虽然病毒载体是目前最有效的载体,但它们的应用依然受限于其承载能力、免疫原性和致癌性[67-68]。
设计的sgRNA与非目标DNA错配,并导致意外的非特异性基因修饰,即为脱靶效应。脱靶效应可能导致序列突变、缺失、重排、免疫反应和癌基因激活等有害事件,严重限制CRISPR/Cas9系统在临床治疗方面的应用[5]。目前主要通过对sgRNA优化、Cas9核酸酶的修饰、其他Cas变体的使用等策略来降低脱靶效应[57]。研究人员发现,健康人体内既有对Cas9蛋白的体液(抗Cas9抗体)免疫反应,也有细胞(抗Cas9 T细胞)免疫反应。由于CRISPR/Cas9系统来源于细菌,它们可以感染人类,因此如何检测和降低Cas9蛋白的免疫原性仍然是该系统临床试验中最重要的挑战之一[69]。此外,CRISPR/Cas9可以诱导p53信号通路的激活,即经过基因编辑的细胞很可能成为潜在的癌症起始细胞,在治疗中额外诱发癌症。如使用CRISPR/Cas9对1 000多条斑马鱼的基因组进行编辑时,第一代和其后代都产生意外的突变,并且这种突变可以遗传[70]。最后,人们担心以生育为目的的基因编辑会永久性地改变人类基因库,因此目前不应尝试将人类基因组编辑用于生殖目的[71]。
