天然海藻二糖中羟基选择性保护技术研究
2022-11-24刘金瀚顾也欣梁观峰
刘金瀚,顾也欣,梁观峰
(1.河南工业大学 化学化工学院,河南郑州 450001;2.上海昱聚科技有限公司,上海 201100;3.复旦大学 化学系,上海 200433)
羟基广泛存在于天然单糖及多糖等物质中,是决定糖类化合物结构和性质的重要官能团[1-3]。羟基同时也是有机合成中重要的官能团,可以进一步转化为醛基、胺基、羧基等多种衍生化官能团。由于其性质活泼,在进行糖类化合物的衍生化反应过程中,往往需要将部分羟基保护起来,尤其是在涉及多官能团复杂分子的修饰或合成中,如何选择性地保护羟基官能团是糖类化合物衍生化的关键步骤,例如在紫杉醇的合成中羟基的保护与脱保护策略[4-5]。羟基的保护主要是将其转化为相应的醚或者酯,一般用于羟基保护的醚有硅醚、甲基醚、苄基醚、烷氧甲基醚及三甲基硅乙基甲基醚等[6-7]。此外,缩醛、缩酮类化合物也是保护羟基的常用方法[8]。
1 材料与方法
1.1 实验原理
在本研究中,首先以甲醇和环己酮为原料高收率地合成了环己酮二甲缩酮[9],具体反应机理如图1所示。然后在有机质子酸的催化下,用环己酮二甲缩酮保护海藻二糖的羟基,通过对反应的优化,得到选择性保护的产物。实验未选用较为多样化的环己酮二乙缩酮的合成方法生成中间产物[10],而是采用环己酮二甲缩酮作为中间产物进行选择性保护,与其他方法相比,收率明显增高,对于后续的羟基选择性实验起到积极影响[11]。
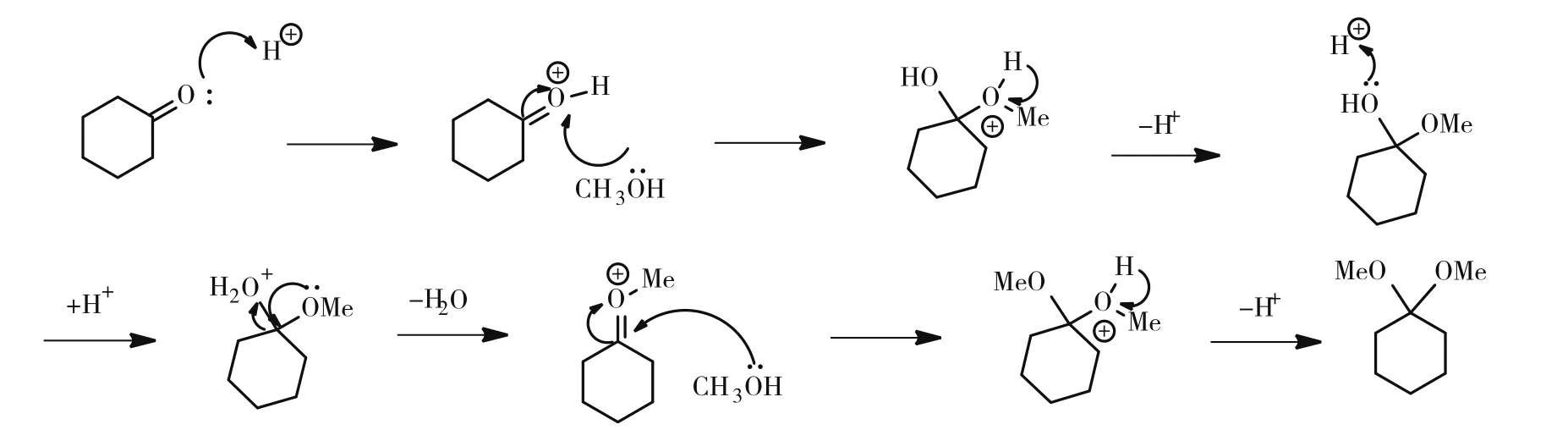
图1 环己酮缩酮化反应机理
1.2 仪器与试剂
二甲基亚砜(DMSO),化学纯,美国Biosharp公司;盐酸(36.0%~38.0%),分析纯,太仓沪试试剂有限公司;无水甲醇、环己酮、海藻糖、碳酸氢钠、石油醚和乙酸乙酯,分析纯,上海泰坦化学有限公司;对甲基苯磺酸,分析纯,上海易恩化学技术有限公司;原甲酸三甲酯(TMOF),分析纯,上海麦克林生化科技有限公司。
AVANCE Ⅲ 400 MHz型核磁共振波谱仪,德国Bruker公司;7820A/5977B型气相色谱-质谱仪,美国Agilent Technologies公司;6890A型气相色谱-氢火焰离子化检测仪,美国Agilent Technologies公司;DB-624毛细管柱(30 m×0.53 mm,3 μm),美国Agilent Technologies公司;FA2204N型电子天平,上海菁海仪器有限公司;DZF-6020A型真空干燥箱,上海力辰仪器科技有限公司。
1.3 实验方法
1.3.1 产物合成
(1)环己酮与甲醇在盐酸及原甲酸三甲酯(TMOF)催化下进行缩酮化反应。设置3组试验,标号为1、2、3,分别固定环己酮物质的量为0.1 mol、0.3 mol、0.5 mol,TMOF 为 0.1 mol,并分别置于装有搅拌子的烧瓶中,将过量的甲醇(1 mol)缓慢滴加至冰水浴的烧瓶中;将0.5 g质量分数为36%的盐酸以及0.1moL的TMOF用10 mL甲醇稀释后,逐滴加入每组混合溶液中(1、2、3号实验中质子酸与环己酮的摩尔比分别为0.139、0.046、0.028);滴加结束后将烧瓶从冰水浴中取出于室温下反应,水浴加热至60 ℃左右旋蒸除去大部分甲醇后待用。
同时进行放大平行实验,标号为4、5;试剂剂量为环己酮0.3 mol,甲醇3 mol,36%盐酸1.5 g,TMOF为0.3 mol即38.1 g。进行相同实验操作,并测量放大实验数据。
(2)环己酮二甲缩酮在有机质子酸的催化下保护海藻二糖的羟基的反应机理,如图2所示[5]。取上述4和5号放大实验得到的环己酮二甲缩酮,加入原环己酮10%物质的量(0.03 mol)的海藻糖,并加入二倍海藻糖质量(20.54 g)的DMSO,将10%海藻糖物质的量(0.517 g)的对甲基苯磺酸用3 mL的DMSO溶解后,逐滴加入反应液中,60 ℃反应3 h;配制饱和NaHCO3溶液,边搅拌边将反应液逐滴加入到NaHCO3溶液中,滴加结束后得到白色沉淀,用超纯水洗涤沉淀3遍,去除质子酸、DMSO及未反应的海藻糖等杂质,55 ℃、真空干燥得到白色粉末状粗产物;将粗产物用体积比为10∶1的石油醚和乙酸乙酯混合溶剂洗涤3遍,常温晾干得到最终产物,称重并记录实验数据。
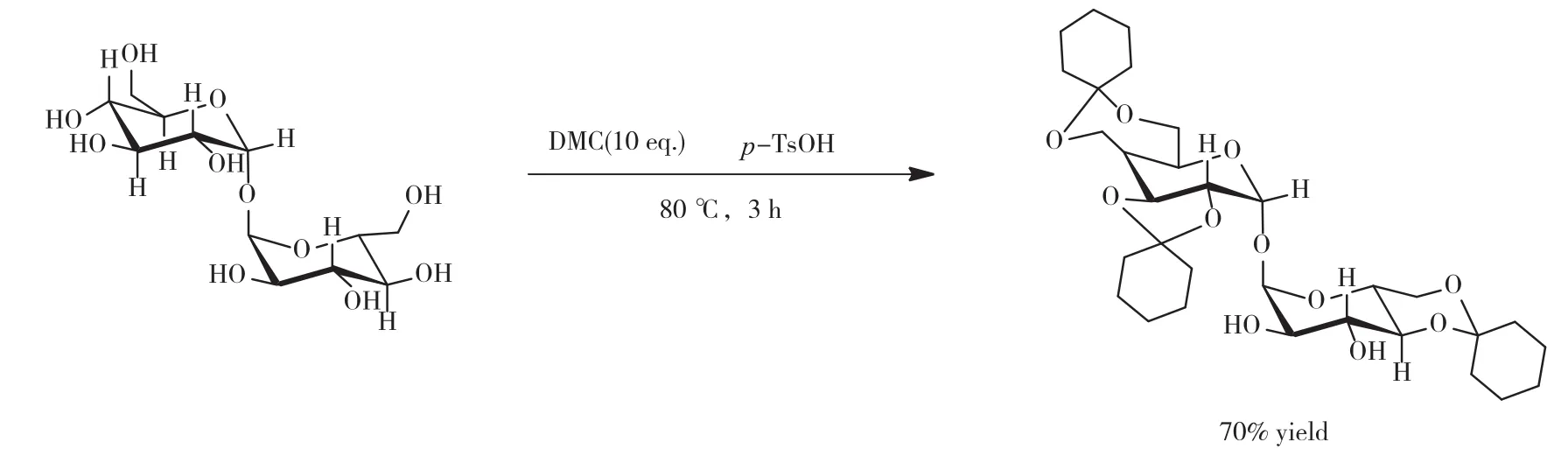
图2 环己酮二甲缩酮在质子酸的催化下保护海藻二糖羟基
1.3.2 表征实验
气相色谱-质谱仪(GC-MS)确定第一步产物的分子结构(定性分析),气相色谱-火焰离子化检测仪(GC-FID)确定转化率和收率(定量分析);第二步反应的产物由核磁共振定性确定。
优化的气相色谱-质谱条件:载气压力为0.02 MPa,载气流量为40 mL/min,氢气压力为0.04 MPa,空气压力为0.08 MPa,气化室温度为250 ℃,检测室温度为160 ℃,柱温为60~250 ℃,加热速率为15 ℃/min,分流比为100∶1。使用电子轰击离子源,全扫描采集模式,扫描范围为40~217 amu。
核磁共振氢谱条件:采用4 mm的双共振探头对白色沉淀物进行采集,魔角旋转转速为10 kHz,1H的共振频率为399.33 MHz。
1.4 数据处理
(1)确定产物。在反应过程中,缩酮化反应中理想状况下1当量环己酮应生成1当量份环己酮二甲缩酮,而在选择性保护过程中,因取代基个数不确定,因此需要确定取代产物。故后续利用色谱图确定取代产物,并计算其相对原子质量。
(2)计算产率。利用公式(1)计算产率。
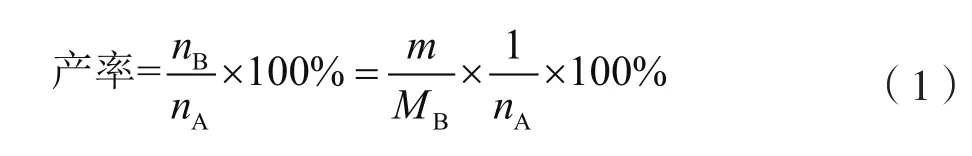
式中:m为需测量产物的质量,g;MB为需测量产物的摩尔质量,g/mol;nB与nA分别为产物和原料的物质的量,mol。
2 结果与分析
2.1 环己酮与甲醇的缩酮反应
环己酮与甲醇发生缩酮化反应,得到无色透明液体,用GC-MS定性分析该产物,结果如图3所示。从图3(a)可以看到,1.0 min、5.1 min、5.4 min和6.3 min有物质峰出现;从图3(b)可知,6.3 min时分离得到相对原子量为144.1的分子,为目标产物——环己酮二甲缩酮(C8H16O2,分子质量为144.115 0);从图3(c)可知,5.4 min时分离得到的副产物杂质相对原子量为112,推测可能发生了脱水与消除反应,生成了含双键及单甲氧基的副产物,如图3(d)所示(C7H12O,分子质量为112)。

图3 缩酮反应产物表征
2.1.1 环己酮浓度对反应产物的影响
质子酸与环己酮的摩尔比例对产物的目标转化率具有影响,表1为3个环己酮浓度下第一步反应产物占比及转化率。由表可知,1号实验组的目标产物转化率最高,达到93.9%,较大程度上反映了第一组反应物的配比有利于反应向右进行,较低浓度的环己酮反应程度较高,于是在第一组的基础上进行了对应环己酮质子酸浓度的放大实验。
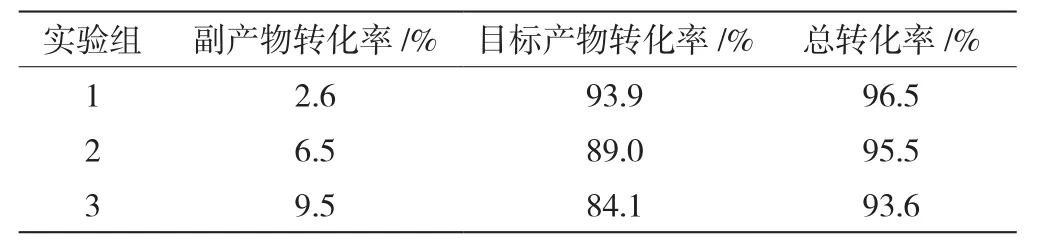
表1 第一步反应产物占比及转化率
2.1.2 放大实验
表2为放大实验中第一步反应产物占比及转化率。两组平行实验的转化率差异较大,5号实验组产生了23.4%的副产物,可能是5号实验组实际冰水浴时间比4号少,体系温度较高,发生消除反应、加剧脱水,使副产物占比增高。

表2 放大实验中第一步反应产物占比及转化率
2.2 海藻糖的选择性羟基保护
第二步得到白色粉末,不溶于水及石油醚,易溶于甲醇和乙醇。海藻糖加入量为10.27 g,理论产物质量为16.0 g,实际得到白色粉末15.1 g,实验产率为44.93%。同时,对第二步的产物进行了核磁共振分析,如图4所示。1H NMR(400 MHz,CDCl3),δ:7.31~7.42(m,1H),6.69~6.75(m,1H),5.01~5.26(d,1H),3.46~4.19(m,2H),1.41~1.84(s,3H),0.81(s,1H)。上述结果初步说明了产物为三取代物。
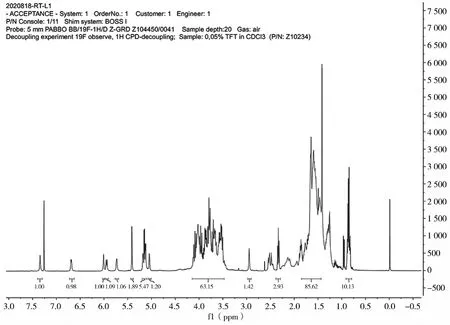
图4 海藻酸羟基保护产物核磁分析谱图
3 结论
在本实验中,首先以甲醇和环己酮为原料高收率地合成了环己酮二甲缩酮,然后在有机质子酸的催化下,用环己酮二甲缩酮保护海藻二糖的羟基,通过对反应的优化,得到三环已酮保护的产物,实现了对海藻糖羟基的选择性保护。此外,通过平行试验,推测出反应中可能存在温度对副反应的影响,即实验过程中应严格监控温度,减少副反应的进行。
