拉考沙胺的合成工艺研究
2022-11-24张之建刘可可刘源
张之建,刘可可,刘源
(上药康丽(常州)药业有限公司,江苏常州 213105)
拉考沙胺化学结构式如图1,化学名为(2R)-2-乙酰氨基-N-苄基-3-甲氧基丙酰胺,其抗癫痫作用被认为是调节钠通道缓慢失活,并在一些随机对照试验已显示其疗效和耐受性。一个单中心、大型队列研究了连续服用拉科酰胺片治疗难治性癫痫的保留率,结果发现1年为62%,2年为45%,3年为35%。服药期间有18%患者报告发作显著减少或发作停止超过6个月,其中4例发作停止超过1年[1]。

图1 拉考沙胺结构式
在过去的数十年里,已经开发了几种拉考沙胺的合成路线。CHOI等[2]以D-丝氨酸为起始物料开发了第一代拉考沙胺的合成路线,如图2所示。该路线主要优势在于通过D-丝氨酸直接引入了手性中心,并且无需对氨基进行保护。但由于氨基未保护,会使得化合物3裸露的氨基与甲基化试剂反应,前两步反应收率仅为27%。第二代拉考沙胺也是由CHOI及其研发团队开发的,但由于氧化银需要5倍反应当量,因此不具备产业化价值。

图2 第一代拉考沙胺合成路线
比利时优时比公司开发了第三代拉考沙胺合成路线,如图3所示[3]。该合成路线第一个优势在于羟基甲基化之前对氨基进行Boc基团保护,避免了氨基甲基化;通过使用相转移催化剂四丁基溴化铵,避免了氧化银的使用,大大降低了生产成本。第二个优势为适当条件下拉考沙胺的总摩尔收率达到了69.8%。但该合成路线中化合物5制备过程中反应温度较为宽泛,手性纯度不稳定,主要原因在于化合物7的手性纯度中异构体含量维持在2.5%左右需要通过不断重结晶来达到异构体要求;并且未对中间体的纯度进行控制,仅控制了目标化合物纯度,因此制备出的拉考沙胺质量不稳定。
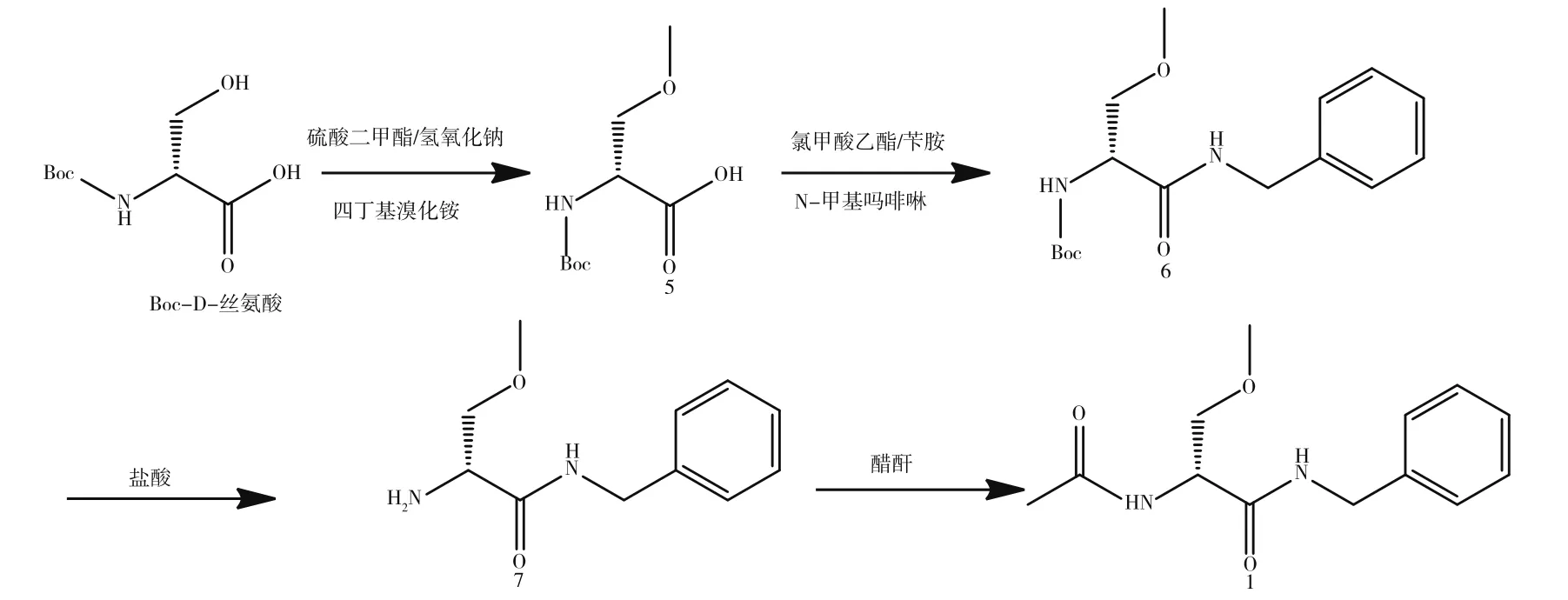
图3 第三代拉考沙胺合成路线
在优时比工艺的基础上,其他的氨基保护基团也有所报道,例如采用苄氧羰基[4]和叔丁基[5]基团对氨基进行保护,但造成反应收率明显降低。其他的与第三代合成反应路线类似的工艺也被开发出来[6-7],但仅是甲基化试剂与乙酰化试剂的改变,且以损失收率的方式获取高纯度的目标化合物,没有实质性的突破。其他与第三代不一样的合成路线也被开发出来[8-15],比如通过拆分或者不对称合成获取手性中心。
考虑到工艺产业化,第三代合成路线仍是目前最有优势的方法,因此本研究团队对第三代拉考沙胺合成路线重新梳理,旨在开发一条提高收率、降低目标化合物质量不稳定性的工艺路线。
1 试剂与仪器
Boc-D-丝氨酸,工业级,常州市华人化工有限公司;硫酸二甲酯,工业级,淄博远辉化工有限责任公司;苄胺,分析纯,上海泰坦科技股份有限公司;氯甲酸乙酯,工业级,徐州市利群化工有限公司;36%盐酸,工业级,常州市海通化工有限公司;L-酒石酸,工业级,常茂生物化学工程股份有限公司;醋酐,工业级,常州市玉宇化工有限公司;拉考沙胺对照品(LCAP1批次产品),上药康丽(常州)药业有限公司。
AUW220D电子天平,日本岛津;C-MAG HP 7加热器,德国艾卡;RV 10 Gigital V旋转蒸发仪,德国艾卡;安捷伦1260液相色谱仪,美国安捷伦。
2 实验步骤及结果
2.1 化合物5的合成
向250 mL三口瓶内加入100 mL甲苯,搅拌下加入10 g Boc-D-丝氨酸,0.52 g四丁基溴化铵,降温至0~10 ℃;控制温度低于10 ℃滴加10 g 20%的氢氧化钠溶液,滴加完毕后加入24.6 g硫酸二甲酯,降温至-5~5 ℃;控制温度低于5 ℃,分批缓慢加入18 g 50%的氢氧化钠水溶液,于0~10 ℃保温搅拌20 h。
保温结束后向反应体系内加入30 mL水,搅拌10 min,静置分相,水相用50%的柠檬酸调节pH值<3.5;分别用50 mL、50 mL、30 mL二氯甲烷萃取水相3次,萃取完毕后将有机相浓缩至干。取样通过HPLC面积归一化法分析,化合物5含量99.7%,最大单杂0.08%,用于下一步制备。
2.2 化合物6的合成
将1.2.1制备的化合物5溶解于80 mL二氯甲烷中,溶解后降温至-10~0 ℃;向反应体系内加入6.7 g氯甲酸乙酯,完成后控制温度-10~5 ℃,滴加7.8 g N-甲基吗啡啉,保温搅拌1 h。保温结束后控制温度-10~-5 ℃滴加苄胺的二氯甲烷溶液(苄胺5.4 g+二氯甲烷20 mL),滴加完成后缓慢升温至10~15 ℃,保温搅拌2 h。
保温结束后向反应体系内加入20 mL水,搅拌10 min,静置分相;有机相用20 mL盐酸(1 mol/L)洗涤1次,静置分相;有机相用20 mL碳酸氢钠溶液(8%)洗涤1次,静置分相;有机相用20 mL水洗涤1次,静置分相,得化合物6的二氯甲烷溶液。取样通过HPLC面积归一化法分析,化合物6含量95.6%,最大单杂1.3%,备用。
2.3 化合物7的L-酒石酸盐合成
将1.2.2制备的化合物6的二氯甲烷溶液降温至0~10 ℃,控制温度10 ℃以下滴加10 g盐酸(36%),滴加完成后缓慢升温至20~25 ℃,保温搅拌4 h。保温结束后向反应体系内加入20 mL水,搅拌10 min,静置分相;有机相用20 mL的水洗涤2次,合并水相;向水相中加入40 mL二氯甲烷,之后用30%的氢氧化钠水溶液调pH至12,静置分相;水相用20 mL二氯甲烷萃取2次,合并有机相,将有机相浓缩至干。
向浓缩物中加入20 mL丙酮,控制温度20~30 ℃滴加L-酒石酸的丙酮溶液(L-酒石酸6.6 g,丙酮15 mL),于20~30 ℃保温搅拌2 h;保温结束后抽滤,滤饼用适量冷的丙酮洗涤,于30~40 ℃下真空干燥,得化合物7的L-酒石酸盐13.1 g,三步总摩尔收率75.1%。取样通过HPLC面积归一化法分析,化合物7的L-酒石酸盐含量99.83%,最大单杂0.05%,异构体0.05%。
2.4 拉考沙胺的合成
向100 mL三口瓶内加入6 g水和3 g化合物7的酒石酸盐,之后降温至10~15 ℃;向反应体系内加入6 g二氯甲烷,控制温度10~15 ℃滴加2.4 g氢氧化钠溶液(30%),静置分相,水相用5 g二氯甲烷萃取2次,合并有机相。
控制温度5~10 ℃缓慢滴加醋酐0.8 g,滴加完成后升温至20~25 ℃,保温搅拌4 h。保温结束后向反应体系内加入3 g水,静置分相,分别用3 g碳酸氢钠水溶液(8%)洗涤有机相1次、3 g水洗涤有机相2次,将二氯甲烷相浓缩至干。
向浓缩物中加入18 g乙酸乙酯,升温至回流溶清,缓慢降温至0~10 ℃,保温搅拌2 h;保温结束后抽滤,滤饼用冷的乙酸乙酯淋洗,于50 ℃下真空干燥,收料1.8 g。参考欧洲药典分析方法,使用标准对照品定位,通过HPLC面积归一化法分析,目标产物含量99.8%以上,异构体含量小于0.1%。以化合物7为基准计算,本步反应摩尔收率85.2%。
3 结论
本研究团队在第三代拉考沙胺合成路线基础上将L-酒石酸这一手性拆分剂引入到合成路线中,化合物7的手性纯度中异构体含量由2.5%左右降低至0.1%以下,大大提高了化合物7的手性纯度;仅需一次乙酸乙酯结晶即可达到欧洲药典(https://pheur.edqm.eu/home)标准要求,减少了对目标化合物拉考沙胺的纯化步骤;以Boc-D-丝氨酸为起始物料计,摩尔收率达到了63%,收率优于第一代拉考沙胺合成工艺路线,工艺稳定性优于第二代拉考沙胺合成工艺路线,对工业化生产具有指导意义。
