二代测序技术在血管性血友病诊断中的应用
2022-11-22周泽平
杨 文,周泽平
(昆明医科大学第二附属医院 血液内科,云南 昆明 650000)
血管性血友病(von Willebrand disease,VWD) 是最常见的常染色体遗传出血性疾病,主要是由于血管性血友病因子(von Willebrand factor, VWF)基因突变所致。国外报道VWD发病率约为1/1 000[1],但我国尚无明确流行病学资料。VWF是血浆中参与凝血过程的大分子多聚体糖蛋白,依据VWF水平和功能可将VWD分为定量异常和定性异常(见图1),定量异常包括1型和3型,定性异常仅包括2型,而2型又被分为2A,2B,2M和2N四种亚型。这三种类型和各亚型之间具有不同的生理病理机制,临床表现、治疗方法也有各自特点。VWD的诊断主要基于阳性家族史、不明原因的出血及异常的实验室检测结果[2-4]。国际血栓与止血协会出血评估工具有助于医生了解病情,但不具备特异性,需要进一步完善实验室检查。而常规的筛查试验,如血细胞计数、活化部分凝血活酶时间和凝血酶原时间等,在大多数VWD患者中可能是正常的。准确诊断VWD还需做以下检查: VWF抗原(von Willebrand factor antigen,VWF:Ag),瑞斯托霉素诱导血小板聚集试验(ristomycin-induced platelet aggregation,RIPA),瑞斯托霉素辅因子活性测定(von Willebrand factor ristocetin cofactor,VWF:RCo),VWF胶原结合活性(VWF collagen binding,VWF:CB),VWF与凝血因子VIII结合能力(VWF and coagulation factor VIII binding activity,VWF:FVIIIB),凝血因子VIII活性(coagulation factor VIII coagulant activity,FVIII:C),VWF多聚体分析,去氨加压素(1-deamino-8-D-arginine vasopressin, DDAVP)试验,VWF前导肽(VWF propeptide, VWFpp)测定和VWF基因测序,但是这其中有很多耗时且技术性强的实验操作在大多数医院并没有常规开展。此外,由于VWF基因突变外显率低、患者临床表现异质性大、实验室检测变异性高及 VWF 水平易受外界因素干扰(如遗传、激素、环境、衰老等),诊断变得更加复杂,甚至导致误诊[5]。尽管传统使用的Sanger等测序法可以识别VWF基因突变位点,有助于诊断,但存在耗时长、成本高、低通量等缺点,并未在临床广泛开展。随着分子诊断技术的发展,二代测序技术(next-generation sequencing,NGS)应运而生,它具有高通量、高灵敏度、价格低廉等众多优点,现已被证明是遗传性疾病基因诊断的有效手段之一[6]。近年来,NGS在VWD诊断中广泛应用,为临床诊疗决策提供了更多可靠的依据。本文就NGS在VWD诊断中的应用进行综述,旨在为临床诊断提供依据。
1 NGS原理
NGS又称高通量测序,是一种新兴的分子诊断技术。现有的二代测序平台包括但不限于Roche/454、Illumina/Solexa GenomeAnalyzer、Polonator、Ion Torrent。不同的平台,测序原理有所不同,但其核心工作原理都是边合成边测序,即用4种不同的荧光分子标记脱氧核糖核苷三磷酸(deoxyribonucleoside triphosphate,dNTP),在DNA分子进行聚合酶链式反应(polymerase chain reaction,PCR)扩增时,每一种新添加的dNTP会释放不同的荧光信号,测序仪通过捕获这些荧光信号,并导入特定计算机软件处理,从而获得待测DNA的序列信息。
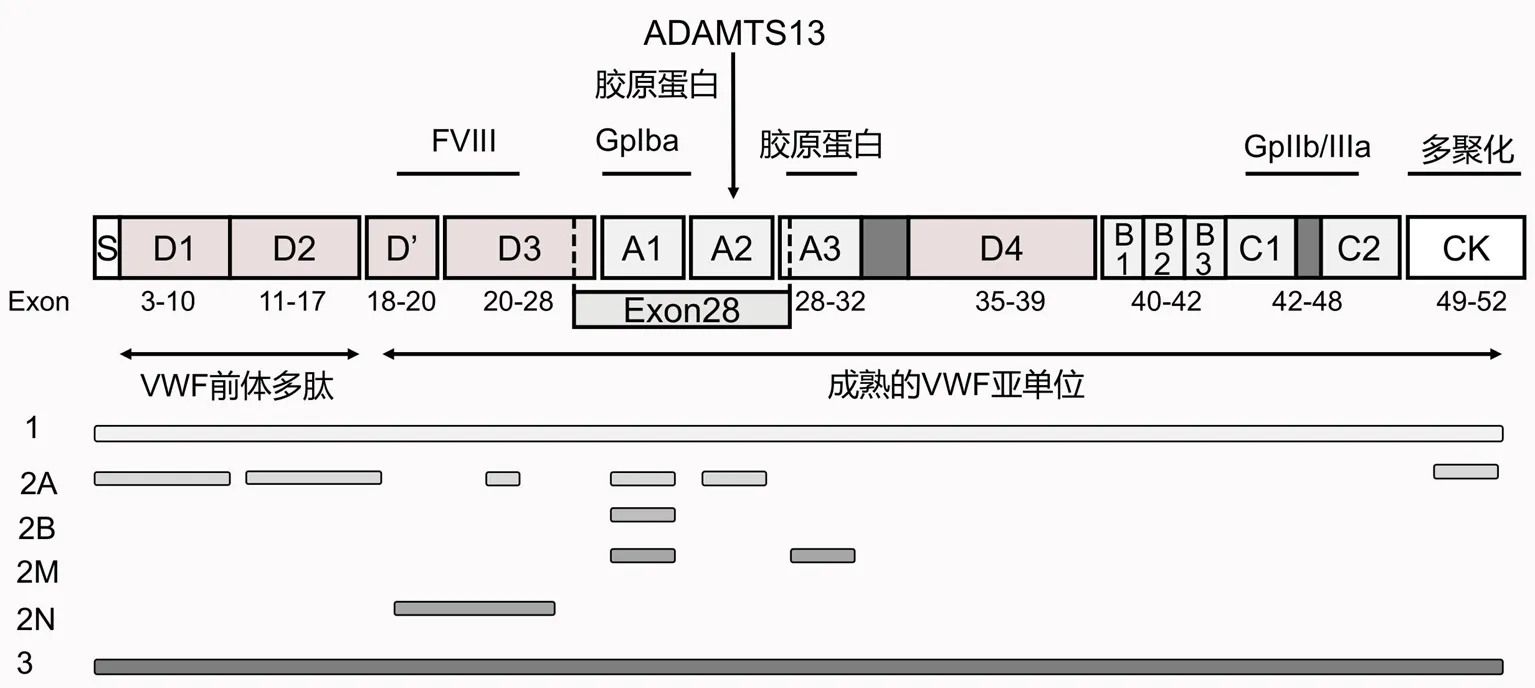
图1 VWF的分子结构与VWD各类型的突变定位
2 NGS在VWD诊断中的应用
2.11型VWD 1型VWD最多见(占75%),为常染色体显性遗传,其特点是血浆VWF水平仅为正常人的20%~50%。1型患者的基因突变位点散布于整个VWF基因,从启动子10到外显子52[7],主要集中在28号外显子,常见的突变形式为错义突变(占75%)。突变体主要影响VWF合成或分泌以及血浆VWF清除效率,如D3区C1149R可以降低VWF的分泌,D4区C2257S可以影响VWF合成,同时,D4区L2207P会导致VWF在细胞内滞留[8]。还有一些突变会使血浆 VWF 清除速率显著加快,如W1144G、I1416N[9]。目前已测序的1型患者突变率为70%[10],剩余30%虽然没有发现基因突变,但这些患者可能涉及到更为复杂的遗传因素,比如内含子及其他遗传位点的突变。
大多数1型患者为单个突变的显性遗传,但由单个突变导致出血的患者仅占5%~10%[11-12]。这表明除遗传缺陷外,还存在其他因素与低水平VWF相关,比如ABO血型就是其中的一种。1987年Gill等[11]就报道了O型血的人比正常人VWF水平低,这对于O型血患者诊断VWD非常重要,因为O型血在1型VWD中出现的比例高达65%[12]。VWFpp位于VWF分子结构的D1和D2区域,通常它与成熟的VWF被等比例地分泌到血液中,但两者的半衰期不同,VWFpp和VWF: Ag比值可以反映体内VWF的合成、分泌及清除状况[13]。VWFpp和VWF: Ag比值升高提示VWF清除增快,这在1C型VWD患者中更加明显(1C型是1型VWD中的特殊亚型)[14]。此外,修饰基因也会影响VWF水平。CLEC4M是一种凝集素受体,以往研究证明它可以清除血浆VWF[15],但最近一项瑞典的研究发现CLEC4M还可以清除血浆中已经结合了FVIII的VWF[16]。STXBP5基因曾被证实其编码的突触融合蛋白结合蛋白-5可通过抑制内皮细胞胞吐导致低水平的VWF。后来Lind-Hallden等[17]又进一步发现了STXBP5基因中的p.N436S变异与1型VWD相关。Sabater-Lleal等[18]还发现了11个可能影响VWF水平的修饰基因:PDHB/PXK/KCTD6,SLC39A8,FCHO2/TMEM171/TNPO1,HLA,GIMAP7/GIMAP4,OR13C5/NIPSNAP,DAB2IP,C2CD4B,RAB5C-KAT2A,TAB1/SYNGR1,ARSA。但是上述新基因在VWD中的功能特征有待进一步研究。
2.22型VWD
2.2.12A型VWD 2A型是2型VWD中最常见的一种,多数为常染色体显性遗传,其特点是高-中分子量VWF多聚体缺失,导致血小板依赖性的功能减弱,大多数患者VWF活性和VWF:Ag比值降低。几乎所有2A型VWD患者都可以检测出异常突变,其中约73%的突变位于28号外显子[19]。2A型VWD与50多种不同的错义突变有关,这些错义突变位于VWF不同的结构域,其发病机制有以下3种:血浆VWF裂解蛋白酶(ADAMTS13)敏感性增强(A1和A2结构域),VWF二聚化或多聚化缺陷(D1-D2和CK结构域),VWF在细胞内滞留(D3结构域)[20]。各结构域的致病机制和热点突变不同(见表1)。
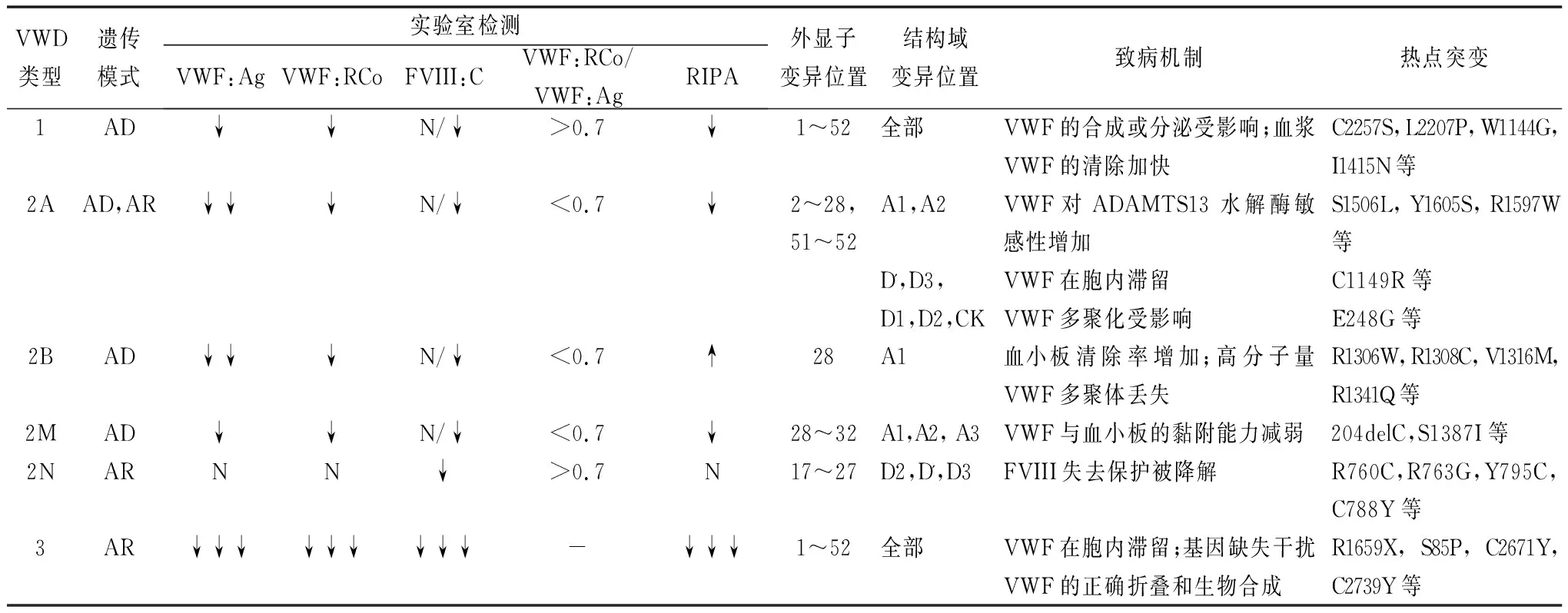
表1 VWD各类型的实验室检测特点、热点突变及致病机制
2.2.22B型VWD 2B型VWD为常染色体显性遗传,此型是由于VWF基因的A1区突变导致VWF与血小板膜表面糖蛋白(glycoprotein,GP)Ib受体亲和性增加所致,这种突变会导致血小板过度聚集并出现消耗性减少,高分子量VWF多聚体降低甚至缺失。低浓度的瑞斯托霉素(<0.5 mg/ml)诱导血小板聚集是2B型VWD的主要特征,同样也是诊断的主要方法之一[21]。2B型VWD与血小板型血管性血友病(platelet-type von Willebrand disease,PT-VWD)有类似的表型,但后者是由于GP1BA基因发生功能性突变,导致血小板膜表面GPIb结合VWF能力增加,通常使用NGS可帮助鉴别两者[22]。目前国际VWF突变数据库(http://www.ragtimedesign.com/vwf/mutation)已发现50多种2B型突变,这些突变都位于28号外显子,且多数突变已被证明具有致病性。几乎所有2B型突变为错义突变,其中R1306W、R1308C、V1316M和R1341Q为热点突变,约占2B型突变的80%[23]。这些突变位点与VWF多聚物减少和A1结构域的结合能力增强有关。Maas等[24]报道了一种特殊类型的2B突变P1266L,此突变与高分子多聚物水平和血小板减少无关。
2.2.32M型VWD 2M型VWD为常染色体显性遗传,其发病机制为A1结构域突变导致对血小板膜表面GPIb的亲和力降低,RIPA减低,但VWF:Ag含量与多聚体分析正常。2M型VWD的突变类型包括错义突变(93%)和缺失突变(7%),其中多数突变发生在28号外显子,其余的位于外显子17,27,30,31和52。临床上2M型很容易误诊为2A型,因为两者的实验室检测结果相似。高分子量VWF的减少是2A型的重要特征,所以全面的表型分析有助于2M型和2A型的鉴别。此外,有研究发现A3结构域的错义突变可使VWF与胶原蛋白的结合能力下降,进而削弱血小板的黏附能力,导致2M型VWD[25]。
2.2.42N型VWD 2N型VWD发病机制为VWF基因D’区和D3区突变,导致VWF结合FVIII的能力减弱,血浆FVIII失去保护而被降解,FVIII水平显著降低(<10%)。目前所观察到的2N型突变大约有60种,约95%为错义突变,主要位于18~20号外显子,另外一些位于4、9、17、24~27外显子。不同于大多数常染色体显性遗传的VWD类型,2N型是隐性遗传,两个突变位点可以是纯合的单一突变、不同的杂合突变、错义突变与无效突变的杂合。不同突变类型可能会影响患者的FVIII水平。Michiels等[26]发现纯合子p.Arg816Trp(R816W)突变通常与临床严重的2N型VWD相关。另一项意大利的研究表明,纯合子2N型VWD患者FVIII水平和VWF和FVIII:Ag比值明显降低,而这些参数在复合杂合子中显示正常或接近正常[27]。Casonato等[28]建议常规检测VWF:FVIIIB,因为这不仅可以帮助可疑2N型VWD的诊断,还能防止误诊。2N型患者的临床表现和实验室检查与轻/中度A型血友病几乎相同,极易混淆,以往主要通过详细的家族遗传史或实验室检测VWF与FVIII结合能力(VWF:FVIIIB)来区分两者,但随着NGS的普及,对VWF基因上的FVIII结合位点进行测序可以做到准确诊断。
2.33型VWD 3型VWD非常罕见(<1%),为常染色体隐性遗传,主要是由于两条VWF等位基因发生纯合或双重杂合突变导致血浆VWF完全缺乏和FVIII活性显著降低(1%~9%),患者通常伴有严重的出血表现。研究表明,除了传统的隐性遗传模式外,大约40%~50%的3型VWD还表现出共显性遗传[29]。国际血栓与止血协会科学标准化委员会的VWF基因突变数据库中已报道了110余种不同的3型突变,包括插入、无义和错义突变以及部分或全部VWF基因缺失。这些突变散布在整个VWF基因,其中28号外显子突变占比最大,最常见的无义突变为R1659X。由于3型VWD患者的VWF基因存在众多突变位点,这也直接导致了此型患者出血严重程度的差异性。加拿大一项对3型VWD患者进行出血评分的研究表明,VWFpp发生突变的患者比其他地方突变的患者出血更严重[29]。相比1型和2型VWD,3型VWD临床表现严重、血浆VWF绝对缺乏、FVIII水平极低,从而更容易诊断。但是完善二代测序仍是必要的,特别是3型患者家庭计划生育小孩时,NGS可以帮助全面了解父母的VWF基因,做到从源头上控制致病基因的传递,防止缺陷儿的出生。
3 小结
VWD的诊断是一个临床难题。尽管通过表型分析足以确定大部分患者的VWD类型,但表型分析不能充分解释部分患者的病情,仍需要进一步完善基因测序。无论是诊断2N型VWD,还是鉴别2B型与2M型或PT-VWD,NGS都发挥出了其明确的优势。3型VWF基因突变位点存在异质性,特别是VWF基因部分或全部缺失时,DNA测序不能提供有效诊断,可能还需要其他的诊断策略,如多重连接依赖式探针扩增技术[30]或cDNA Southern印迹法寻找异常的杂交特征。1型VWD突变位点在整个VWF基因的广泛分布限制了NGS的效果,但在与其他类型的VWD鉴别时,基因测序仍然是排除潜在1C型或2型的最快方法。综上,NGS高通量、快速、灵敏的特性补充了传统测序方法的不足,在诊断和鉴别VWD方面有很高的临床应用价值。未来,随着测序技术的进一步发展,有望实现真正的个体化医疗,造福更多的患者。
