溶质载体SLC家族在非酒精性脂肪性肝病中的研究进展
2022-11-21汤志全石丽熊晶
汤志全,石丽,熊晶
综 述
溶质载体SLC家族在非酒精性脂肪性肝病中的研究进展
汤志全,石丽,熊晶
中国药科大学药学院,南京 210009
非酒精性脂肪肝病(non-alcoholic fatty liver disease,NAFLD)与肥胖症和2型糖尿病密切相关,是代谢综合征的组成部分之一。由于NAFLD发病机制的复杂性,目前尚无针对性的药物治疗。溶质载体(solute carrier,SLC)转运蛋白与多种代谢性疾病有关,在肝脏中具有丰富表达,参与多种营养物质和代谢产物的转运,调节营养供应、代谢转换、能量平衡和氧化应激等,调控肝脏生理功能。尤为重要的是,其中部分SLC转运体已经成为药物开发的新靶标。本文重点阐述SLC在营养物质和肝脏代谢产物转运中的作用及其与NAFLD的相关性,并揭示SLC作为NAFLD新药研发潜在靶标的可能性,以期为治疗NAFLD提供新的选择。
非酒精性脂肪性肝病;溶质载体SLC家族;糖脂代谢
代谢综合征是由肥胖、高血糖、高甘油三酯、低高密度脂蛋白胆固醇水平和高血压等组成的一组临床表现,往往表现为多种代谢紊乱集于一身,且肥胖可成为这些代谢紊乱的共同诱因,目前靶向药物治疗很大一部分集中在肥胖与T2D。NAFLD与肥胖症、T2D密切相关,是代谢综合征的组成部分之一[1,2]。NAFLD是指除长期大量饮酒和其他明确的肝损伤因素外所引起的,以甘油三酯为主的脂质在肝细胞中蓄积为病理特征的肝脏代谢性疾病,疾病谱包括单纯性脂肪变性、非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH)、肝硬化和肝细胞癌。其中,NASH以炎症、肝细胞损伤和肝纤维化为特征,可进展为肝硬化和肝细胞癌[3]。流行病学调查显示,NAFLD全球成人患病率超过25%,并且已成为中国最常见的肝脏疾病,呈现出年轻化的发病趋势。预计到2030年底,我国NAFLD患者将达到3.14亿[4]。NASH患者中约15%可发展为肝硬化、肝细胞癌等终末期肝病,后者是肝移植的主要原因,对人类生命健康造成威胁[5]。因此,深入探究NASH发生发展的分子机制对于靶向NASH药物的研发和NASH患者的药物干预治疗均具有重要意义。
SLC家族包含400多种转运蛋白,这些转运蛋白介导离子、氨基酸、核苷酸和糖等小分子物质跨生物膜流入和流出。SLC介导被动和继发性主动转运,其中沿电化学梯度进行的被动转运不需要耗能[6],而继发性主动转运与离子的顺电化学梯度转运相耦合,成为SLC底物运输的能量来源。介导物质和驱动离子向同一方向运输的称为共转运体(同向转运体),而交换体(反向转运体)则向相反方向转移物质。例如,共转运体SLC5A1、SLC5A2分别介导上皮细胞和近端小管细胞Na+摄取和葡萄糖重吸收[7],交换体SLC16A1编码的质子相关单羧酸转运蛋白负责单羧酸如乳酸和丙酮酸在质膜上的交换[8]。SLC转运蛋白在肝脏、肾脏、大脑和肠道等代谢器官中具有较高的表达水平,可以感知和响应细胞外营养物质的浓度、激素水平和能量状态的波动,与糖脂代谢关系密切,在肝脏的功能稳态调节中发挥关键作用。目前已知,超过80种SLC转运蛋白与人类疾病有关,包括肥胖和T2D[9]。值得注意的是,肝脏脂肪变性合并代谢异常易引起并发症,而代谢综合征又与NASH进程密切相关,两者相互促进。当前NAFLD相关药物研究主要聚焦于NASH治疗新策略的研发,目前在研新药有200余种,其中我国在研新药30余种[10]。然而,SLC蛋白如何受细胞内外信号调节从而转运代谢底物,促进细胞代谢并影响NAFLD的机制还有待更深入的研究。本文主要综述了代谢底物转运体SLC在肝脏糖脂代谢和NAFLD发生发展过程中的相关研究和潜在价值,揭示了SLC转运蛋白作为NAFLD药物靶标的理论依据。
1 SLC基因家族成员的定位及生物学作用
SLC介导各种底物的跨膜运输,如无机离子、氨基酸、脂肪酸、神经递质和糖类等,在细胞生理功能的维持中发挥重要作用。NAFLD相关SLC家族成员的细胞内定位,生理功能和表达情况总结见表1。
2 SLC与NAFLD的相关性
2.1 单糖转运体SLC2/SLC5
葡萄糖是机体的主要能量来源,SLC2A家族葡萄糖转运体(glucose transporter member,GLUT)和SLC5A家族Na+-葡萄糖共转运体(sodium/ glucose cotransporter member,SGLT)参与处理葡萄糖和其他己糖的吸收、分布、排泄和代谢,维持全身葡萄糖稳态[11]。几乎所有SLC2A都在肝脏中表达,其中SLC2A1、SLC2A2、SLC2A4、SLC2A5、SLC2A8和SLC2A9在肝脏中的表达尤其丰富。胰腺细胞通过SLC2A2感知血糖浓度升高,并增加胰岛素分泌。在胰岛素作用下,SLC2A4转移到质膜,进一步促进胰岛素与其受体结合,促进葡萄糖转运到骨骼肌、脂肪组织和心脏细胞中加以利用。同时,SLC2A2和SLC2A4也介导肝脏对葡萄糖的摄取,而这两种转运蛋白表达异常会降低葡萄糖摄取和利用效率,引起机体代偿性地分泌过多胰岛素而产生高胰岛素血症和高血糖症,破坏肝脏胰岛素和葡萄糖稳态。
↑表示在NAFLD中上调,↓表示在NAFLD中下调。
SGLT以Na+与溶质共转运为特征,介导葡萄糖从肾小管腔重新吸收到细胞内,由ATP酶建立的Na+浓度梯度驱动,可回收超过90%经肾小球滤过的葡萄糖[12]。其中,SGLT1(又称为SLC5A1)和SGLT2(又称为SLC5A2)是上皮细胞葡萄糖转运的重要转运体,也是SLC5A家族的主要成员。SLC5A2负责肾小管系统大部分的葡萄糖重吸收,随后,SLC5A1重吸收其余滤过的葡萄糖。基因突变导致葡萄糖和半乳糖吸收不良,而基因突变与糖尿病有关,该基因突变小鼠表现出体重减轻、糖尿、多尿,但血糖正常,没有胰岛素抵抗或肾功能障碍的迹象[7]。其他SLC5A家族成员如SLC5A10也可能具有调控代谢稳态的潜力。
2.1.1 葡萄糖转运体SLC2A1/2/4/9和SLC5A1/2
SLC2A1是调节己糖转运的主要SLC2A家族成员,具有非常广泛的底物特异性,可以运输多种醛糖[13]。在胚胎发育过程中,SLC2A1缺陷导致葡萄糖摄取减少,激活细胞凋亡,诱发肝坏死[14]。一致的是,表达水平在NAFLD患者肝脏中降低,而且敲除的THLE2细胞内活性氧水平升高、油酸诱导的脂滴增多[15]。同样,SLC2A2介导葡萄糖通过肝细胞膜的双向转运,在NASH肝脏中表达降低[16]。体外研究表明,高糖培养的HepG2细胞中SLC2A2表达降低而磷酸烯醇丙酮酸羧激酶-1和6-磷酸葡萄糖激酶升高,并伴随胰岛素抵抗和脂质沉积[17]。因此,SLC2A1和SLC2A2可能通过促进肝脏糖酵解和抑制活性氧水平而阻碍NAFLD进展。
SLC2A4介导骨骼肌和脂肪组织中胰岛素调控的葡萄糖转运进入细胞,肌肉和脂肪细胞含有的一种特殊细胞器GLUT4储存囊泡(GLUT4 storage vesicle,GSV)受胰岛素刺激后,在细胞表面融合并将SLC2A4蛋白插入质膜。基因缺陷小鼠因骨骼肌和脂肪组织葡萄糖摄入减少而引发高血糖症[18],更重要的是,胰岛素对该基因的调控不仅存在于特定的细胞类型如脂肪和肌肉细胞,也存在于肝脏细胞[19]。因此,SLC2A4在肌肉、脂肪和肝脏细胞中增加胰岛素对葡萄糖的摄取,可能通过维持全身葡萄糖稳态来缓解NAFLD。此外,尿酸转运体SLC2A9将葡萄糖和果糖转运至细胞内,维持血浆尿酸和葡萄糖正常水平,SLC2A9肝脏特异性失活小鼠表现出严重的高尿酸血症[20],而高水平尿酸可诱导小鼠肝脏脂肪异常积累[21],进而可能增加患NAFLD风险。
由SLC5A1和SLC5A2介导的葡萄糖重吸收促进NAFLD的发展过程,且目前研究主要集中在SLC5A2特异性抑制剂对NAFLD的治疗作用。SLC5A2抑制可通过下调脂质合成相关蛋白,上调脂肪酸氧化相关基因(比如PPARα和CPT1α),保护L02和HepG2免受棕榈酸诱导的细胞内脂质堆积所造成的毒性[22]。基因在小鼠肝脏中有表达,尤其在肝巨噬细胞和T细胞中表达水平较高[23]。SLC5A2抑制通过改善全身胰岛素抵抗,导致体重降低,同时减少白色脂肪组织[24],也可通过促进肝脏脂肪酸氧化、抑制脂肪合成来减少肝脏甘油二酯、甘油三酯和胆固醇含量,以减轻肝脏脂毒性[25]。临床研究表明,SLC5A2抑制通过降低血糖、肝脏脂肪含量和体重来改善NAFLD患者肝脏功能。临床前实验和临床研究都表明SLC5A2抑制通过改善糖脂代谢,减少肝细胞氧化应激、炎症和细胞凋亡,从而减轻NAFLD程度[26]。
2.1.2 果糖转运体SLC2A2/5/8和SLC5A10
果糖特异性细胞膜转运蛋白SLC2A5在十二指肠、小肠和肾脏组织中高表达,在肝脏中表达水平相对较低。果糖经小肠上皮细胞SLC2A5转运后进入血液循环,由肝细胞SLC2A2摄取后经过一系列酶促反应生成二羟基丙酮磷酸和甘油醛-3-磷酸,促进糖异生过程而导致血糖升高。果糖代谢中间产物也可通过激活碳水化合物反应元件结合蛋白(carbohydrate response element-binding protein,ChREBP)和固醇调节元件结合蛋白-1C(sterol regulatory element-binding protein 1C,SREBP1C)促进肝细胞脂肪合成[27]。SLC2A5与小肠中果糖运输高度相关,但也可能有助于肝细胞对果糖的摄取,果糖摄入过量也与肝脏表达增加有关,可引起线粒体氧化应激和功能障碍,促进肝脏炎症反应及NAFLD进程[28]。此外,果糖摄入过量也可引起内质网应激,促进肝细胞凋亡[29]。相比于仅高脂饮食,高果糖高脂饮食模型有最接近人类NAFLD的病变特征,SLC2A5对于果糖摄取至关重要,并且通过升高血糖、增加肝脏脂肪含量来诱导NAFLD。
SLC2A8促进葡萄糖和果糖转运进入细胞内,在肝脏中高度表达。SLC2A8缺陷的雌鼠表现出肝果糖首过代谢受损,敲除显著阻断HepG2的果糖摄取,这表明果糖转运到肝细胞部分是由SLC2A8介导的[30]。缺陷小鼠能够抵抗高果糖饮食诱导的葡萄糖耐受不良和高血脂症[31]。有趣的是,雌鼠比雄鼠更易受高果糖饮食影响而诱导肝脏脂肪含量增加,而且肝脏果糖激酶在雌性大鼠中显著地被果糖摄入所诱导,而在雄性大鼠中则没有[32]。进一步研究表明,靶向敲除肝脏可通过增强脂肪酸氧化,显著降低脂肪酸含量,减轻果糖诱导的内质网应激和肝脏炎症,对果糖诱导的NASH雌鼠肝损伤和炎症具有预防作用[33]。
SLC5A10介导肾近端小管细胞的果糖重吸收,其缺陷可引起尿果糖排泄,减少果糖吸收。矛盾的是,缺陷小鼠尿液中有大量果糖排泄,伴随着血浆甘油三酯和附睾脂肪水平的降低,但表现出更严重的果糖诱导的肝脂肪变性以及空腹高胰岛素血症,可能是因为SLC5A10缺陷导致SREBP-1c前体活化入核并增加肝脏脂肪合成[34],提示SLC5A10在NAFLD疾病进程中的作用有待更深入的研究。
2.2 硫酸盐/羧酸盐转运体SLC13
SLC13家族Na+偶联阴离子转运体介导三羧酸循环中间产物如柠檬酸、琥珀酸和α-酮戊二酸等由质膜转运到细胞内,调节这些代谢物在血浆、尿液和组织中的水平,从而调节糖脂代谢[35]。SLC13转运蛋白分为两类:一类SLC13A1 (NaS1)和SLC13A4 (NaS2)运输硫酸盐,另一类SLC13A2 (NaC1/NaDC1)、SLC13A3(NaC3/NaDC3)和SLC13A5 (NaC2/NaCT)运输羧酸盐,这些转运蛋白主要在肝、肾、小肠和脑中表达[36]。
2.2.1 羧酸盐转运体NaDC1、NaDC3和NaCT
编码的Na+/二羧酸共转运体1 (Na+/ dicarboxylate cotransporter 1, NaDC1)在肾脏和小肠中表达尤高,分别从尿液和饮食中重吸收琥珀酸、α-酮戊二酸和柠檬酸等三羧酸循环中间产物,其重要功能是介导柠檬酸的重吸收,维持血液中酸碱平衡稳态[37]。柠檬酸盐通过抑制磷酸果糖激酶-1来抑制糖酵解,并通过刺激果糖-1,6-双磷酸酶来激活糖异生,因此,细胞内柠檬酸盐水平的降低会刺激糖酵解并抑制糖异生[38]。柠檬酸盐在胞浆中的代谢物乙酰辅酶A对于脂肪酸合成和蛋白质乙酰化都很重要,且促进巨噬细胞活化[39]。高脂饮食小鼠肝脏基因表达下调,而二甲双胍能上调饮食诱导肥胖小鼠肝脏的表达[40],推测SLC13A2可能通过转运柠檬酸进入细胞内,升高细胞内柠檬酸盐水平,抑制肝脏糖酵解和促进脂肪合成,可能加重NAFLD。
编码的Na+/二羧酸共转运体3 (Na+/ dicarboxylate cotransporter 3, NaDC3)可转运α-酮戊二酸作为肝细胞内谷氨酰胺合成的底物[41],谷氨酰胺可降低高脂饮食诱导的肝脏氧化应激程度,抑制肝脏脂肪变性,对NAFLD有一定的抑制作用[42]。此外,肥胖合并2型糖尿病大鼠肝脏中基因表达上调[43],临床研究表明,NAFLD合并肥胖患者血浆α-酮戊二酸水平升高[44],提示SLC13A3可能转运α-酮戊二酸进入细胞内,促进谷氨酰胺合成的同时抑制肝脏脂肪变性而减轻NAFLD。
编码的Na+/柠檬酸共转运体(Na+/ citrate cotransporter, NaCT)对柠檬酸的亲和力最高,其主要功能是将血液循环中的柠檬酸转运进入细胞内,参与利用细胞外柠檬酸盐合成脂肪酸和胆固醇,促进脂质累积[45]。在肥胖、胰岛素抵抗的NAFLD患者的肝脏样本中表达显著增加,并伴随肝脏脂肪变性。基因敲除小鼠可抵抗高脂饮食和衰老诱导的肥胖、肝脏脂肪变性和胰岛素抵抗[46],同样,基因敲除的人肝癌细胞系HepG2也显示出柠檬酸盐摄取及其分解代谢受损,脂质水平降低[47],提示SLC13A5具有类似于SLC13A2的抑制NAFLD作用。
2.2.2 硫酸盐转运体NaS1
编码的Na+/硫酸盐共转运体1 (Na+/ sulfate cotransporter, NaS1)可促进硫酸盐在小肠的吸收和肾脏的重吸收,从而维持硫酸盐血浆水平稳态。缺陷被证明与肝损伤血清酶学指标(血清转氨酶丙氨酸氨基转移酶和天冬氨酸氨基转移酶)的水平升高密切相关[35],而该基因缺失的小鼠表现出肝脏脂肪、血浆胆固醇和低密度脂蛋白增多,肝脏糖原含量减少[48],提示SLC13A1缺失可能抑制肝脏糖异生并增加脂质合成以诱发NAFLD。
2.3 单羧酸转运体SLC16
SLC16由单羧酸转运体(monocarboxylate transporter,MCT)家族的14个成员组成,在细胞营养物质的运输、新陈代谢和酸碱平衡的调节中发挥重要作用。SLC16A1,SLC16A4,SLC16A10,SLC16A11和SLC16A13通过参与L-乳酸、丙酮酸、短链脂肪酸和其他单羧酸类物质在各种组织中的质子依赖性转运,与糖脂的动态调节有关,可能影响NAFLD过程中糖、脂和氨基酸代谢[49]。
2.3.1 SLC16A1和SLC16A4
SLC16A1介导单羧酸盐和酮体在肝细胞膜上的转运。糖酵解产生的乳酸是肝脏糖异生和脂肪生成的底物,肝脏通过SLC16A1和SLC16A4促进乳酸的排出,维持细胞内乳酸平衡,乳酸是TCA循环中的主要碳源,因此也是能量生成的主要碳源[50]。另外,糖酵解过程中产生的丙酮酸也是TCA循环中的主要碳源。在禁食情况下或生酮饮食时,肝脏酮体β-羟丁酸酯和乙酰乙酸酯大量生成,由SLC16A1转运出肝脏,可为大脑、心脏和骨骼肌提供能量[51]。基因敲除小鼠可能通过抑制乳酸和丙酮酸外排,促进糖酵解过程和TCA循环,抵抗高脂饮食诱导的肥胖,减轻胰岛素抵抗和肝脏脂肪变性[52]。SLC16A4将单羧酸盐转入细胞内,在肥胖、糖尿病和肾脏损伤患者中表达升高[53],且高表达的NASH患者易转变为更严重的肝癌[54],可能是SLC16A4将过多的乳酸盐、丙酮酸盐和酮体转运进入细胞内,脂质积累异常所致。
2.3.2 SLC16A10
SLC16A10介导肝脏上皮细胞内芳香族氨基酸流出,维持体内循环中和肝脏中氨基酸水平[55]。NASH小鼠肝脏中下调,而且可能是NASH肝脏中芳香族氨基酸,尤其是酪氨酸和苯丙氨酸浓度增加的潜在原因[56]。因此,MCT10将氨基酸代谢与NAFLD联系起来,可能部分通过调节血浆氨基酸水平和肝脏胰岛素水平抑制NAFLD进展[57]。
2.3.3 SLC16A11
SLC16A11在肝脏中高表达且主要定位于内质网,参与肝脏脂质代谢过程。NAFLD的主要病理学特征是肝脏异常的脂质积累,破坏肝细胞中内质网蛋白稳态,导致炎症损伤甚至细胞死亡[58]。人类原代肝细胞敲除后胞内酰基肉碱、二酰甘油和三酰甘油水平显著增加,肝细胞以极低密度脂蛋白形式分泌的三酰甘油胞外水平也增加[59]。与之一致的是,NAFLD患者血浆和肝脏酰基肉碱、二酰甘油和三酰甘油水平增加[60]。然而,敲除小鼠无显著代谢缺陷,但在基因敲除小鼠的肝脏中重组2型糖尿病患者的突变体会导致更多的甘油三酯积累,诱导胰岛素抵抗,而重组野生型不会造成甘油三酯积累和胰岛素抵抗[61]。相反,敲低通过抑制血浆和肝脏甘油三酯蓄积,进而改善高脂饮食喂养小鼠的葡萄糖耐量和肝脏胰岛素信号传导[62]。因此,SLC16A11在肝脏脂质代谢中的作用十分复杂,可能通过影响肝脏中甘油三酯积累与NAFLD相联系,其在NAFLD中的具体作用及机制值得进一步的研究。
2.3.4 SLC16A13
SLC16A13在肝脏中高表达且主要定位于高尔基体,转运乳酸进入细胞内。高脂饮食喂养的基因敲除小鼠可能通过减少肝细胞内乳酸供应,导致AMP活化蛋白激酶(AMP-activated protein kinase, AMPK)激活,肝细胞线粒体氧化呼吸功能增强,胰岛素敏感性增加,肝脏脂肪堆积减少[63]。此外,PPARα作为肝脏脂代谢的关键调节因子,其激动剂可上调小鼠小肠和肝脏中基因的表达[64]。因此,SLC16A13可能通过细胞内过量乳酸和脂肪堆积推动NAFLD进程。
2.4 有机阳离子和阴离子转运体SLC22
SLC22转运蛋白家族在肝脏、肾脏和肠道中高表达,可介导肝细胞中外源性有机离子的摄取,根据底物性质分为3个亚家族:有机阳离子转运体(organic cation transporter, OCT)、有机阴离子转运体(organic anion transporter, OAT)和有机阳离子/肉碱转运体(organic cation/carnitine transporter, OCTN)。OCTs分布于各类组织,在亲水性药物的肝脏吸收中起重要作用[65]。OATs亚家族约占SLC22转运蛋白家族的一半,通过调节肝脏信号分子和关键代谢物水平而在细胞间通讯中发挥作用[66],包括SLC22A7/9/12。OCTN是膜转运蛋白的一个小亚家族,可介导人肾脏和肝脏对肉碱的重吸收,从而升高血浆和肝脏组织肉碱水平,促进线粒体脂肪酸β氧化[67]。
2.4.1 肉碱转运体SLC22A5
SLC22A5将肝脏细胞膜上L-肉碱转运至细胞内,而且长链脂肪酸依赖于与L-肉碱的酯化反应形成乙酰肉碱,以便从细胞质运输到线粒体基质进行氧化和能量产生[68]。L-肉碱还有另一个重要作用,即通过形成乙酰肉碱被转运出线粒体,缓冲线粒体中过量的乙酰辅酶A[69]。SLC22A5缺陷引起原发性肉碱缺乏症,患者血清和肝细胞内L-肉碱水平较低,导致脂肪酸氧化受阻,患者因完全依赖葡萄糖进行能量代谢而出现低血糖[70]。原发性肉碱缺乏症患者与NAFLD患者有相似的肝脏病理特征,表现为肝脏脂肪堆积、ALT和AST水平升高[71]。因此,SLC22A5通过提高肝细胞内肉碱含量而促进脂肪酸氧化,进而抑制NAFLD。
2.4.2 有机阴离子转运体SLC22A7/9/12
SLC22A7在肝脏和肾脏中高表达,对环核苷酸例如环鸟苷单磷酸(cyclic guanosine monophosphate, cGMP)有较高的底物亲和力[72],可能在调节细胞信号转导过程中发挥作用。此外,烟酸和烟酰胺是烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide, NAD+)的生物合成前体,主要由SLC22A7转运进入肝细胞[73]。NAD+是一种重要的能量代谢氧化还原因子,也是还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide Phosphate, NADPH)的前体,NADPH是抗氧化防御系统的重要组成部分。据此推测,SLC22A7转运NAD+前体进入细胞内,可能改善肝脏线粒体功能,降低氧化应激,抑制肝星状细胞激活,阻遏NASH进程[74]。
SLC22A9是SLC22家族有机阴离子转运体成员中的第一个肝脏特异性转运体,只在肝脏中表达,并定位于肝细胞的窦状膜上。SLC22A9介导S2细胞的丁酸摄取和硫酸雌酮外排[75],且丁酸钠已被证实可以改善高脂饮食诱导的肝脏炎症和肝功能指标ALT和AST,降低肝脏甘油三酯和胆固醇水平,减轻NAFLD[76]。此外,HNF-1α与基因启动子区域结合,促进该基因表达,靶向敲低HNF-1α降低了Huh7细胞中表达[77];HNF-1通过抑制PPARγ和SREBP1/2等脂肪生成相关基因的表达来抑制脂质合成代谢,从而缓解NAFLD进展[78],推测可能通过增强HNF-1α对脂质合成代谢的抑制作用而降低NAFLD风险。
SLC22A12在肝脏和棕色脂肪组织中表达,并在HFD小鼠棕色脂肪组织中表达上调而导致棕色脂肪白色化和脂质积累,并增加了棕色脂肪组织活性氧的产生。的表达抑制可以减轻HFD诱导的肥胖小鼠肝脂肪变性,促进富含脂质的BAT重新褐变来改善胰岛素抵抗,可能是伴有NAFLD的胰岛素抵抗个体的新治疗靶点[79]。
2.5 线粒体转运体SLC25
SLC25家族是SLC转运蛋白家族中最大的家族,由53个成员组成[80]。SLC25成员在其运输底物的性质和大小、运输方式(单向、同向或反向)和驱动力方面差异很大。SLC25转运体主要定位于线粒体内膜,介导氨基酸、脂肪酸、辅因子、无机离子和核苷酸转运,参与多种代谢途径,包括TCA循环、脂肪酸氧化和合成、糖酵解和糖异生、氨基酸分解等代谢过程[81]。
核苷酸转运体包括线粒体ADP/ATP载体SLC25A4、SLC25A5、SLC25A6、SLC25A31高表达于肝脏、肾脏和脂肪组织,将ADP输入到线粒体基质,并将产生的ATP输出到细胞质中,为细胞代谢提供能量[82]。SLC25与肝脏糖脂代谢相关研究相对缺乏,在NAFLD调控中可能有一定的潜力。
解偶联蛋白(uncoupling protein, UCP)调节线粒体膜电位、能量消耗、活性氧产生和细胞氧化还原状态。UCP介导质子从膜间隙进入线粒体基质,从而破坏质子梯度、降低膜电位[83]。UCP的表达和功能与代谢性疾病密切相关,UCP失调或功能障碍可能通过增加氧化应激诱导的肝脏损伤而加剧NAFLD进程[84]。
2.5.1 解耦联蛋白SLC25A7/8/9
SLC25A7在棕色脂肪组织中高表达,解偶联氧化磷酸化和ATP产生,将能量转化为热量。棕色脂肪组织产热在动物模型肥胖的发生中起关键作用,而SLC25A7功能障碍诱导小鼠模型肥胖的发生[85]。缺陷小鼠肝脏表现出细胞外琥珀酸含量升高,通过激活肝脏星状细胞和巨噬细胞琥珀酸受体1来驱动炎症,加速NAFLD进展[86]。白色脂肪细胞中SLC25A7受寒冷、药物、营养和内源性刺激诱导,促进白色脂肪棕色化,另一方面,可能增强NAFLD小鼠肝脏线粒体呼吸功能,改善肝脏脂肪变性和胰岛素抵抗[87]。小鼠棕色脂肪组织中脂滴外壳蛋白Perilipin 5在暴露于寒冷环境中显著增加,维持线粒体嵴结构完整性和促进线粒体呼吸功能,而SLC25A7可能通过增强Perilipin 5对线粒体功能的促进作用,改善全身糖耐量和高脂饮食诱导的肝脂肪变性[88]。因此,SLC25A7的线粒体解偶联作用对于维持全身能量稳态和治疗肥胖相关NAFLD具有重要意义。
SLC25A8通过抑制线粒体中活性氧产生而发挥抗氧化功能,对于维持胰岛细胞的功能至关重要[89]。糖尿病小鼠胰岛β细胞中SLC25A8活性降低会增加ATP的产生和胰岛素分泌,但同时增加的活性氧阻碍胰岛β细胞分泌胰岛素[90]。此外,肥胖小鼠胰岛细胞中SLC25A8一方面抑制β细胞的葡萄糖依赖型胰岛素分泌,另一方面增加能量消耗,从而降低肥胖的风险[91]。SLC25A8增加胰岛素分泌,可能通过降低血糖浓度及促进肝脏胰岛素功能以缓解NAFLD。
SLC25A9与SLC25A8功能相似,同时可介导脂肪酸诱导的解偶联,增强脂肪酸β氧化相关的中链酰基辅酶A-脱氢酶和过氧化物酶体增殖物激活受体-γ共激活因子-1α的基因表达,改善线粒体功能[92],并可能通过抑制肝脏脂毒性而减轻NAFLD。
2.5.2 三羧酸循环中间产物转运体SLC25A1/ 10/11/13/20/24
线粒体柠檬酸盐转运体SLC25A1将柠檬酸从线粒体基质转运到胞浆,且在NASH肝脏中高表达。肝脏特异性敲除可以降低细胞内柠檬酸水平,减弱PPARγ信号传导,并抑制脂质合成和糖异生基因的表达,改善糖耐量,减少肝脏炎性巨噬细胞浸润,同时显著减轻由高脂肪饮食引起的肥胖[93]。
SLC25A10主要在白色脂肪组织中表达,其次在肝脏中表达,通过线粒体内膜将苹果酸盐转运进入线粒体,将柠檬酸盐从线粒体转运至胞浆,脂肪组织或肝脏细胞浆中的脂肪合成由柠檬酸盐转运开始,肥胖小鼠白色脂肪组织中表达显著增加,以柠檬酸盐为原料合成脂肪[94]。抑制SLC25A10则显著减少了HepG2和3T3-L1细胞中柠檬酸从线粒体到胞浆的运输,降低了ACC1表达和丙二酰辅酶A水平,抑制脂肪合成[95];过表达导致3T3-L1线粒体超极化而产生活性氧[96]。另一方面,琥珀酸经过从线粒体基质转运到胞质,作用于琥珀酸受体1并通过抑制cAMP-磷酸化激素敏感脂肪酶途径来抑制脂肪分解。白色脂肪细胞特异性敲除导致脂肪细胞脂解增强,并促进HFD诱导的小鼠肝脂毒性和全身胰岛素抵抗,而脂肪细胞特异性过表达通过减少肝脏游离脂肪酸的摄取而减轻肝脂毒性,同时改善葡萄糖耐量和胰岛素敏感性[97]。SLC25A10可能主要通过抑制肝脏脂肪合成而作为治疗NAFLD的靶标。
SLC25A11参与苹果酸-天冬氨酸穿梭,部分介导分离的大鼠肝脏线粒体摄取谷胱甘肽,以及H4IIE的谷胱甘肽从细胞质转运到线粒体,抵抗过氧化氢诱导的细胞毒性[98];此外,SLC25A11介导α-酮戊二酸和其他二羧酸盐的交换[99],将α-酮戊二酸从线粒体转移到胞浆,α-酮戊二酸通过抑制相关限速酶直接抑制肝脏糖异生,也可以进入细胞核内,与基因启动子区域结合,且肝脏特异性敲除可消除α-酮戊二酸对PEPCK、G6Pase和FBP蛋白表达和活性的抑制作用[100],提示SLC25A11可能通过介导肝脏细胞中α-酮戊二酸和谷胱甘肽穿过线粒体进行交换,减少肝细胞线粒体氧化应激并抑制糖异生而减轻NAFLD症状。
2.5.3 天冬氨酸-谷氨酸转运体SLC25A13
SLC25A13介导细胞浆谷氨酸转运至线粒体,天冬氨酸从线粒体转运至细胞浆,天冬氨酸是尿素、蛋白质合成,乳酸糖异生以及细胞内NADH氧化所必需的[101,102]。作为肝脏中唯一的天冬氨酸-谷氨酸转运体,SLC25A13缺陷降低肝脏精氨酸琥珀酸合成酶的活性,导致高氨血症[103],基因功能缺陷患者表现出肝脂肪变性和脂肪性肝炎[104]。SLC25A13对肝脏脂肪的调控机制尚不明确,可能通过增加细胞浆中天冬氨酸来源的草酰乙酸,促进糖酵解,随后生成的苹果酸经SLC25A11转运至线粒体促进TCA循环,抑制NAFLD进展。
2.5.4 肉碱转运体SLC25A20
SLC25A20介导胞浆酰基肉碱进入线粒体及线粒体内游离肉碱转移到胞浆,胞浆中肉碱棕榈酰基转移酶1将酰基从乙酰辅酶A转移到肉碱,通过SLC25A20将酰基肉碱转移至线粒体,然后通过肉碱棕榈酰基转移酶2将酰基从酰基肉碱转移到线粒体内的辅酶A,生成的乙酰辅酶A在线粒体中经过β氧化,为机体代谢提供能量[105]。SLC25A20促进肝脏脂肪酸氧化,改变肝脏脂代谢谱,可能抑制NAFLD过程。
2.5.5 ATP-镁/磷酸盐转运体SLC25A24
SLC25A24介导ATP-镁和磷酸盐在线粒体基质和胞浆之间的交换,以及腺嘌呤核苷酸通过线粒体内膜的摄取或流出[106]。当细胞溶质钙水平升高时,SLC25A24将腺嘌呤核苷酸输入线粒体基质,一方面控制线粒体基质腺嘌呤核苷酸水平以响应细胞能量需求,另一方面可能通过促进线粒体基质中磷酸钙沉淀物形成,从而缓冲线粒体基质中钙水平[107]。在应激条件下,腺嘌呤核苷酸进出线粒体对于维持呼吸功能和防止钙诱导的线粒体内膜通透性增加是必要的。高脂饮食诱导小鼠白色脂肪组织和肝脏中基因表达水平升高,该基因敲除小鼠可能通过激活肝细胞内钙离子信号通路,促进氧化分解代谢,进而抵抗HFD诱导的肥胖,表现为体重和白色脂肪组织重量明显降低[108]。
2.6 脂肪酸转运体SLC27
SLC27脂肪酸转运体(fatty acid transport protein,FATP)可存在于细胞膜和细胞质,可作为长链脂肪酸(long chain fatty acids,LCFA)的直接转运体,或作为通过酰基辅酶A合成酶(acyl coenzyme A synthetase,ACS)活性作用于LCFA的酶。ACS催化LCFA转化为酰基辅酶A硫代酯,从而激活LCFA。然后,激活的LCFA可被细胞用于许多代谢过程,如脂肪酸合成、氧化和磷脂合成[109]。与SLC2A类似,SLC27的细胞内定位是动态的,SLC27可以响应胰岛素信号而从细胞质转移到质膜[110],具有摄取和活化LCFA的双重功能。
SLC27A2主要在肝脏和肾脏表达,过表达可增加肝脏脂肪酸摄取和ACS活性[111],肝脏特异性SLC27A2缺失的小鼠肝细胞对LCFA的摄取减少,过氧化物酶体中的ACS活性降低,且SLC27A2缺失能够抑制高脂饮食诱导的肝脂肪变性并改善胰岛素敏感性,可能是治疗NAFLD的新靶点[112]。
SLC27A5仅在肝脏中表达,特别是在肝细胞的基底膜中作为脂肪酸转运蛋白发挥作用。肝脏敲除导致小鼠肝脏甘油三酯和脂肪酸含量降低[113],且通过减少食物摄入量和增加能量消耗而降低高脂饮食导致的体重增加[114],降低肝脏三酯含量和血糖水平。SLC27A5在早期NAFLD患者中的表达增加,抑制SLC27A5可能成为治疗NAFLD的一种新方法。
2.7 锌转运体
SLC30家族中最主要的成员基因的产物锌转运体8(Zinc Transporter 8, ZnT8)表达在胰腺β细胞的胰岛素分泌颗粒膜上,将锌离子从细胞质输送到胰岛素分泌颗粒中,为胰岛素合成提供锌,并且维持胰岛素的成熟、储存和稳定性。与胰岛素协同分泌的锌通过抑制网格蛋白依赖性胰岛素内吞作用来抑制肝脏胰岛素清除,因此β细胞特异性敲除小鼠的胰岛素清除能力增强,外周血胰岛素水平降低[115]。HFD喂养的全身基因敲除小鼠表现出明显增加的肥胖、高血糖、胰岛素抵抗和葡萄糖不耐受;而β细胞特异性敲除小鼠则表现出高血糖、胰岛素合成分泌不足和葡萄糖不耐受,其体重与对照组相似,表明仅β细胞中的不会显著增加体重,可能因为也表达在胰腺α细胞并抑制β细胞功能[116]。适度降低ZnT8活性可以降低β细胞锌分泌,增强肝脏胰岛素清除率;相反,ZnT8活性严重降低可能增加β细胞胰岛素分泌[117]。因此,针对ZnT8的药物干预可能产生较为复杂的结果,体现在对肝脏胰岛素水平和葡萄糖稳态的调控方面,同时对于NAFLD的治疗也有一定的提示作用。
3 SLC介导治疗NAFLD的天然产物或药物
目前SLC家族成员参与的NAFLD治疗主要集中在葡萄糖转运体,另外,三羧酸循环底物转运体也占很大比例,部分代表性天然产物或药物见表2。其中,恩格列净、PF-06649298和L-肉碱作为底物可直接与SLC结合以调控相应的转运体活性,为其他SLC靶向药物的研发提供参考和经验;而其他天然产物或药物通过信号传导途径间接影响SLC的表达和活性,有助于深入研究SLC的上游调控过程,为SLC作为NAFLD治疗药物靶点提供完整和详细的理论依据。
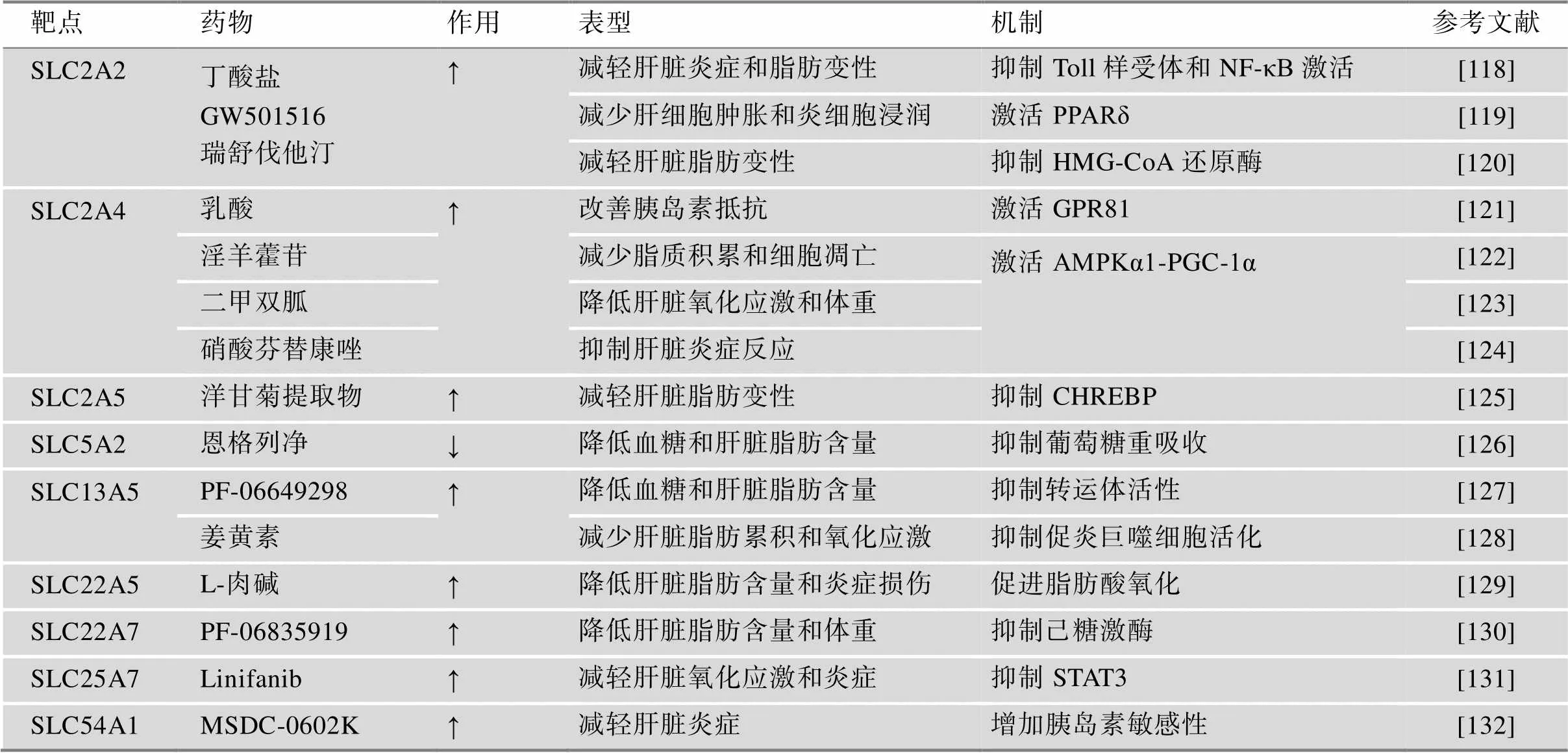
表2 SLC介导治疗NAFLD的天然产物或药物
↑表示上调SLC,↓表示下调SLC。
针对NAFLD的已上市和在研药物中,PPAR激动剂占很大比例,且部分通过调控SLC表达而改善疾病进展。全球仅有印度批准上市首款也是唯一一款NASH治疗药物Saroglitazar,是PPARα/γ的双重激动剂,减少肝脏和血浆中甘油三酯,增强胰岛素敏感性,缓解NASH患者代谢异常[133]。已进入Ⅲ期临床试验的NASH在研新药中,Lanifibranor是一种非选择性PPAR受体激动剂,可同时激活α、δ、γ3种受体,促进葡萄糖的利用,减少肝脏中炎症因子的表达和内质网应激肝脏中的炎症反应[134]。Aramchol是一种胆酸与花生酸的结合物,抑制人类原代肝细胞脂肪合成关键酶SCD1,同时上调保护性基因PPARγ的表达,减轻肝脏炎症反应,为Aramchol用于NASH患者的临床治疗提供了理论依据[135,136]。值得注意的是,PPAR激动剂的广泛激动效应在发挥治疗作用的同时也不可避免地引发非预期结果,给药物研发带来困难和挑战,而SLC作为糖、脂、氨基酸代谢物转运体,其靶向设计药物可直接通过改变代谢物水平以改善代谢紊乱,对于NAFLD的防治具有重要意义。
4 结语与展望
NAFLD与肥胖、T2D联系密切,然而目前NAFLD与SLC的相关研究还不够深入,因此本综述主要从两方面介绍NAFLD与SLC的关系:一方面总结现有研究SLC家族成员在NAFLD的作用及机制;另一方面总结参与到肥胖、T2D中SLC的作用机制,寻找与NAFLD相关的糖脂代谢表型和信号通路,推测其他尚未研究的SLC在NAFLD中可能扮演的角色。同时需要注意的是,针对肥胖或T2D的有效防治也可能有利于NAFLD的防治。
人类代谢性疾病相关的SLC家族转运蛋白中,SLC5A2是唯一已经确证的治疗代谢性疾病的SLC特异性靶点,有4种抑制剂已获FDA批准用于2型糖尿病的临床治疗,可以降低葡萄糖水平、体重、血压和肝脏脂肪含量,同时对于NAFLD的治疗具有潜在有益作用。尽管已有文献报道多种SLC家族成员参与糖脂代谢过程(图1),调控代谢性疾病进展,且与肝脏脂肪变性、炎症反应有关,但在肝脏代谢中的作用及机制还有待深入研究。同时,缺乏结构表征也阻碍了靶向SLC小分子化合物的设计,这也是未来其作为NAFLD防治靶点需要攻克的难点。然而,单一靶点的药物治疗效果有限,结合SLC家族在糖、脂转运中的底物多样性,针对不同发病机制和转运体设计多靶点药物将有助于从代谢物转运的角度理解NAFLD的发病机制,为药物研发提供更详细和有力的理论依据。

图1 SLC参与调控糖脂代谢过程
[1] Samson SL, Garber AJ. Metabolic syndrome., 2014, 43(1): 1–23.
[2] Lemieux I, Després JP. Metabolic syndrome: past, present and future., 2020, 12(11): 3501.
[3] Povsic M, Wong OY, Perry R, Bottomley J. A structured literature review of the epidemiology and disease burden of non-alcoholic steatohepatitis (NASH)., 2019, 36(7): 1574–1594.
[4] Zhou JH, Zhou F, Wang WX, Zhang XJ, Ji YX, Zhang P, She ZG, Zhu LH, Cai JJ, Li HL. Epidemiological features of NAFLD from 1999 to 2018 in China., 2020, 71(5): 1851–1864.
[5] Mundi MS, Velapati S, Patel J, Kellogg TA, Abu Dayyeh BK, Hurt RT. Evolution of NAFLD and its management., 2020, 35(1): 72–84.
[6] Sonveaux P, Maechler P, Martinou JC. Channels and transporters in cell metabolism., 2016, 1863(10): 2359–2361.
[7] Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors., 2018, 61(10): 2079–2086.
[8] Sivaprakasam S, Bhutia YD, Yang SP, Ganapathy V. Short-chain fatty acid transporters: role in colonic homeostasis., 2017, 8(1): 299–314.
[9] Zhang Y, Zhang YP, Sun K, Meng ZY, Chen LG. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development., 2019, 11(1): 1–13.
[10] Chitturi S, Wong VWS, Chan WK, Wong GLH, Wong SKH, Sollano J, Ni YH, Liu CJ, Lin YC, Lesmana LA, Kim SU, Hashimoto E, Hamaguchi M, Goh KL, Fan JG, Duseja A, Dan YY, Chawla Y, Farrell G, Chan HLY. The Asia-Pacific working party on non-alcoholic fatty liver disease guidelines 2017-Part 2: Management and special groups., 2018, 33(1): 86–98.
[11] Chadt A, Al-Hasani H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease., 2020, 472(9): 1273–1298.
[12] Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule., 2011, 22(1): 104–112.
[13] Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family., 1996, 16: 235–256.
[14] Heilig CW, Saunders T, Brosius FC, Moley K, Heilig K, Baggs R, Guo LR, Conner D. Glucose transporter-1- deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy., 2003, 100(26): 15613–15618.
[15] Vazquez-Chantada M, Gonzalez-Lahera A, Martinez- Arranz I, Garcia-Monzon C, Regueiro MM, Garcia- Rodriguez JL, Schlangen KA, Mendibil I, Rodriguez- Ezpeleta N, Lozano JJ, Banasik K, Justesen JM, Joergensen T, Witte DR, Lauritzen T, Hansen T, Pedersen O, Veyrie N, Clement K, Tordjman J, Tran A, Le Marchand-Brustel Y, Buque X, Aspichueta P, Echevarria-Uraga JJ, Martin-Duce A, Caballeria J, Gual P, Castro A, Mato JM, Martinez-Chantar ML, Aransay AM. Solute carrier family 2 member 1 is involved in the development of nonalcoholic fatty liver disease., 2013, 57(2): 505–514.
[16] Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter., 2002, 524(1-3): 199–203.
[17] Su RC, Lad A, Breidenbach JD, Blomquist TM, Gunning WT, Dube P, Kleinhenz AL, Malhotra D, Haller ST, Kennedy DJ. Hyperglycemia induces key genetic and phenotypic changes in human liver epithelial HepG2 cells which parallel the Leprdb/J mouse model of non- alcoholic fatty liver disease (NAFLD)., 2019, 14(12): e0225604.
[18] Wallberg-Henriksson H, Zierath JR. GLUT4: a key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review)., 2001, 18(3): 205–211.
[19] Bryant NJ, Gould GW. Insulin stimulated GLUT4 translocation—Size is not everything!, 2020, 65: 28–34.
[20] Preitner F, Bonny O, Laverrière A, Rotman S, Firsov D, Da Costa A, Metref S, Thorens B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy., 2009, 106(36): 15501–15506.
[21] Xie D, Zhao HR, Lu JM, He FR, Liu WD, Yu W, Wang Q, Hisatome I, Yamamoto T, Koyama H, Cheng JD. High uric acid induces liver fat accumulation via ROS/JNK/AP-1 signaling., 2021, 320(6): E1032–E1043.
[22] Soret PA, Magusto J, Housset C, Gautheron J. In vitro and in vivo models of non-alcoholic fatty liver disease: a critical appraisal., 2020, 10(1): 36.
[23] Shiba K, Tsuchiya K, Komiya C, Miyachi Y, Mori K, Shimazu N, Yamaguchi S, Ogasawara N, Katoh M, Itoh M, Suganami T, Ogawa Y. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH., 2018, 8(1): 2362.
[24] Han T, Fan YJ, Gao J, Fatima M, Zhang YL, Ding YM, Bai L, Wang CX. Sodium glucose cotransporter 2 inhibitor dapagliflozin depressed adiposity and ameliorated hepatic steatosis in high-fat diet induced obese mice., 2021, 10(1): 446–455.
[25] Meng ZY, Liu XH, Li T, Fang T, Cheng Y, Han LP, Sun B, Chen LM. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/ autophagy pathway., 2021, 94: 107492.
[26] Androutsakos T, Nasiri-Ansari N, Bakasis AD, Kyrou I, Efstathopoulos E, Randeva HS, Kassi E. SGLT-2 inhibitors in NAFLD: expanding their role beyond diabetes and cardioprotection., 2022, 23(6): 3107.
[27] Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome., 2010, 7(5): 251–264.
[28] Basaranoglu M, Basaranoglu G, Bugianesi E. Carbohydrate intake and nonalcoholic fatty liver disease: fructose as a weapon of mass destruction., 2015, 4(2): 109–116.
[29] Herman MA, Birnbaum MJ. Molecular aspects of fructose metabolism and metabolic disease., 2021, 33(12): 2329–2354.
[30] DeBosch BJ, Chen Zj, Saben JL, Finck BN, Moley KH. Glucose transporter 8 (GLUT8) mediates fructose- induced de novo lipogenesis and macrosteatosis., 2014, 289(16): 10989–10998.
[31] DeBosch BJ, Chen Zj, Finck BN, Chi M, Moley KH. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice., 2013, 27(11): 1887–1896.
[32] Hyer MM, Dyer SK, Kloster A, Adrees A, Taetzsch T, Feaster J, Valdez G, Neigh GN. Sex modifies the consequences of extended fructose consumption on liver health, motor function, and physiological damage in rats., 2019, 317(6): R903–R911.
[33] Novelle MG, Bravo SB, Deshons M, Iglesias C, García-Vence M, Annells R, da Silva Lima N, Nogueiras R, Fernandez-Rojo MA, Diéguez C, Romero-Picó A. Impact of liver-specific GLUT8 silencing on fructose- induced inflammation and omega oxidation., 2021, 24(2): 102071.
[34] Fukuzawa T, Fukazawa M, Ueda O, Shimada H, Kito A, Kakefuda M, Kawase Y, Wada NA, Goto C, Fukushima N, Jishage KI, Honda K, King GL, Kawabe Y. SGLT5 reabsorbs fructose in the kidney but its deficiency paradoxically exacerbates hepatic steatosis induced by fructose., 2013, 8(2): e56681.
[35] Pajor AM. Molecular properties of the SLC13 family of dicarboxylate and sulfate transporters., 2006, 451(5): 597–605.
[36] Bergeron MJ, Clémençon B, Hediger MA, Markovich D. SLC13 family of Na+-coupled di- and tri-carboxylate/ sulfate transporters., 2013, 34(2-3): 299–312.
[37] Markovich D, Murer H. The SLC13 gene family of sodium sulphate/carboxylate cotransporters., 2004, 447(5): 594–602.
[38] Monchi M. Citrate pathophysiology and metabolism., 2017, 56(1): 28–30.
[39] Williams NC, O'Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation., 2018, 9: 141.
[40] Le JM, Fu Y, Han QQ, Wei XD, Ji HL, Chen YF, Wang QY, Pi PX, Li JL, Lin XJ, Zhang XY, Zhang Y, Ye JP. Restoration of mRNA expression of solute carrier proteins in liver of diet-induced obese mice by metformin., 2021, 12: 720784.
[41] Hagos Y, Schley G, Schödel J, Krick W, Burckhardt G, Willam C, Burckhardt BC. α-Ketoglutarate-related inhibitors of HIF prolyl hydroxylases are substrates of renal organic anion transporters 1 (OAT1) and 4 (OAT4)., 2012, 464(4): 367–374.
[42] Lin ZH, Cai FF, Lin N, Ye JL, Zheng QQ, Ding GS. Effects of glutamine on oxidative stress and nuclear factor-κB expression in the livers of rats with nonalcoholic fatty liver disease., 2014, 7(2): 365–370.
[43] Li YL, Chen D, Xu CM, Zhao Q, Ma YG, Zhao SL, Chen CY. Glycolipid metabolism and liver transcriptomic analysis of the therapeutic effects of pressed degreased walnut meal extracts on type 2 diabetes mellitus rats., 2020, 11(6): 5538–5552.
[44] Aragonès G, Auguet T, Berlanga A, Guiu-Jurado E, Martinez S, Armengol S, Sabench F, Ras R, Hernandez M, Aguilar C, Colom J, Sirvent JJ, Del Castillo D, Richart C. Increased circulating levels of alpha- ketoglutarate in morbidly obese women with non- alcoholic fatty liver disease., 2016, 11(4): e0154601.
[45] Inoue K, Zhuang LN, Maddox DM, Smith SB, Ganapathy V. Human NaCT, the ortholog of Drosophila Indy, as a novel target for lithium action.,2003, 374(1).
[46] Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, Zhang DY, Hsiao JJ, Martin-Montalvo A, Fischer-Rosinsky A, Spranger J, Pfeiffer AF, Jordan J, Fromm MF, König J, Lieske S, Carmean CM, Frederick DW, Weismann D, Knauf F, Irusta PM, De Cabo R, Helfand SL, Samuel VT, Shulman GI. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice., 2011, 14(2): 184–195.
[47] Hu T, Huang WL, Li ZH, Kane MA, Zhang L, Huang SM, Wang HB. Comparative proteomic analysis of SLC13A5 knockdown reveals elevated ketogenesis and enhanced cellular toxic response to chemotherapeutic agents in HepG2 cells., 2020, 402: 115117.
[48] Dawson PA, Gardiner B, Grimmond S, Markovich D. Transcriptional profile reveals altered hepatic lipid and cholesterol metabolism in hyposulfatemic NaS1 null mice., 2006, 26(2): 116–124.
[49] Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE, Morris ME. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease., 2020, 72(2): 466–485.
[50] Halestrap AP, Wilson MC. The monocarboxylate transporter family—role and regulation., 2012, 64(2): 109–119.
[51] Martini T, Ripperger JA, Chavan R, Stumpe M, Netzahualcoyotzi C, Pellerin L, Albrecht U. The hepatic monocarboxylate transporter 1 (MCT1) contributes to the regulation of food anticipation in mice., 2021, 12: 665476.
[52] Droździk M, Szeląg-Pieniek S, Grzegółkowska J, Łapczuk-Romańska J, Post M, Domagała P, Miętkiewski J, Oswald S, Kurzawski M. Monocarboxylate transporter 1 (MCT1) in liver pathology., 2020, 21(5): 1606.
[53] Lokman FE, Seman NA, Ismail AAS, Yaacob NA, Mustafa N, Khir ASM, Hussein Z, Wan Mohamud WN. Gene expression profiling in ethnic Malays with type 2 diabetes mellitus, with and without diabetic nephropathy., 2011, 24(6): 778–789.
[54] Granja SC, Longatto-Filho A, de Campos PB, Oliveira CP, Stefano JT, Martins-Filho SN, Chagas AL, Herman P, D'Albuquerque LC, Reis Alvares-da-Silva M, Carrilho FJ, Baltazar F, Alves VAF. Non-alcoholic fatty liver disease-related hepatocellular carcinoma: immunohistochemical assessment of markers of cancer cell metabolism., 2022, 89(3): 157–165.
[55] Mariotta L, Ramadan T, Singer D, Guetg A, Herzog B, Stoeger C, Palacín M, Lahoutte T, Camargo SMR, Verrey F. T-type amino acid transporter TAT1 (Slc16a10) is essential for extracellular aromatic amino acid homeostasis control., 2012, 590(24): 6413– 6424.
[56] Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, Lehman-McKeeman LD, Vaillancourt RR, Cherrington NJ. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease., 2015, 47(3): 603–615.
[57] Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M, Bugianesi E, Gastaldelli A. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance., 2018, 67(1): 145–158.
[58] Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease., 2018, 69(4): 927–947.
[59] Rusu V, Hoch E, Mercader JM, Tenen DE, Gymrek M, Hartigan CR, DeRan M, von Grotthuss M, Fontanillas P, Spooner A, Guzman G, Deik AA, Pierce KA, Dennis C, Clish CB, Carr SA, Wagner BK, Schenone M, Ng MCY, Chen BH, MEDIA Consortium, SIGMA T2D Consortium, Centeno-Cruz F, Zerrweck C, Orozco L, Altshuler DM, Schreiber SL, Florez JC, Jacobs SBR, Lander ES. Type 2 diabetes variants disrupt function of SLC16A11 through two distinct mechanisms., 2017, 170(1): 199– 212.e20.
[60] Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease., 2020, 42: 101092.
[61] Zhao YX, Feng ZH, Zhang YX, Sun YM, Chen Y, Liu X, Li S, Zhou T, Chen L, Wei Y, Ma D, Lui KO, Ying H, Chen Y, Ding Q. Gain-of-function mutations of SLC16A11 contribute to the pathogenesis of type 2 diabetes., 2019, 26(4): 884–892.e4.
[62] Zhang T, Qi ZT, Wang HY, Ding SZ. Adeno-associated virus-mediated knockdown of SLC16A11 improves glucose tolerance and hepatic insulin signaling in high fat diet-fed mice., 2021, 129(2): 104–111.
[63] Schumann T, König J, von Loeffelholz C, Vatner DF, Zhang DY, Perry RJ, Bernier M, Chami J, Henke C, Kurzbach A, El-Agroudy NN, Willmes DM, Pesta D, de Cabo R, O Sullivan JF, Simon E, Shulman GI, Hamilton BS, Birkenfeld AL. Deletion of the diabetes candidate gene Slc16a13 in mice attenuates diet-induced ectopic lipid accumulation and insulin resistance., 2021, 4(1): 826.
[64] Hirai T, Fukui Y, Motojima K. PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine., 2007, 30(11): 2185–2190.
[65] Koepsell H. Organic cation transporters in health and disease., 2020, 72(1): 253–319.
[66] Nigam SK, Bush KT, Martovetsky G, Ahn SY, Liu HC, Richard E, Bhatnagar V, Wu W. The organic anion transporter (OAT) family: a systems biology perspective., 2015, 95(1): 83–123.
[67] Pochini L, Galluccio M, Scalise M, Console L, Indiveri C. OCTN: a small transporter subfamily with great relevance to human pathophysiology, drug discovery, and diagnostics., 2019, 24(2): 89–110.
[68] Tamai I. Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: SLC22A4, SLC22A5 and SLC22A21)., 2013, 34(1): 29–44.
[69] Rinaldo P, Cowan TM, Matern D. Acylcarnitine profile analysis., 2008, 10(2): 151-156.
[70] Chapoy PR, Angelini C, Brown WJ, Stiff JE, Shug AL, Cederbaum SD. Systemic carnitine deficiency—a treatable inherited lipid-storage disease presenting as Reye's syndrome., 1980, 303(24): 1389–1394.
[71] Deswal S, Bijarnia-Mahay S, Manocha V, Hara K, Shigematsu Y, Saxena R, Verma IC. Primary carnitine deficiency—a rare treatable cause of cardiomyopathy and massive hepatomegaly., 2017, 84(1): 83–85.
[72] Cropp CD, Komori T, Shima JE, Urban TJ, Yee SW, More SS, Giacomini KM. Organic anion transporter 2 (SLC22A7) is a facilitative transporter of cGMP., 2008, 73(4): 1151–1158.
[73] Mathialagan S, Bi YA, Costales C, Kalgutkar AS, Rodrigues AD, Varma MVS. Nicotinic acid transport into human liver involves organic anion transporter 2 (SLC22A7)., 2020, 174: 113829.
[74] Dall M, Hassing AS, Treebak JT. NAD+and NAFLD—caution, causality and careful optimism., 2022, 600(5): 1135–1154.
[75] Shin HJ, Anzai N, Enomoto A, He X, Kim DK, Endou H, Kanai Y. Novel liver-specific organic anion transporter OAT7 that operates the exchange of sulfate conjugates for short chain fatty acid butyrate., 2007, 45(4): 1046–1055.
[76] Zhou D, Pan Q, Xin FZ, Zhang RN, He CX, Chen GY, Liu C, Chen YW, Fan JG. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier., 2017, 23(1): 60–75.
[77] Klein K, Jüngst C, Mwinyi J, Stieger B, Krempler F, Patsch W, Eloranta JJ, Kullak-Ublick GA. The human organic anion transporter genes OAT5 and OAT7 are transactivated by hepatocyte nuclear factor-1α (HNF-1α)., 2010, 78(6): 1079–1087.
[78] Liu F, Zhu X, Jiang XP, Li S, Lv YC. Transcriptional control by HNF-1: emerging evidence showing its role in lipid metabolism and lipid metabolism disorders., 2021, 9(5): 1248–1257.
[79] Tanaka Y, Nagoshi T, Takahashi H, Oi YH, Yoshii A, Kimura H, Ito K, Kashiwagi Y, Tanaka TD, Yoshimura M. URAT1-selective inhibition ameliorates insulin resistance by attenuating diet-induced hepatic steatosis and brown adipose tissue whitening in mice., 2022, 55: 101411.
[80] Ruprecht JJ, Kunji ERS. The SLC25 mitochondrial carrier family: structure and mechanism., 2020, 45(3): 244–258.
[81] Kunji ERS, King MS, Ruprecht JJ, Thangaratnarajah C. The SLC25 carrier family: important transport proteins in mitochondrial physiology and pathology., 2020, 35(5): 302–327.
[82] Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution., 2005, 579(3): 633–637.
[83] Kunji ERS, Robinson AJ. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers., 2010, 20(4): 440–447.
[84] Monteiro BS, Freire-Brito L, Carrageta DF, Oliveira PF, Alves MG. Mitochondrial uncoupling proteins (UCPs) as key modulators of ROS homeostasis: a crosstalk between diabesity and male infertility?, 2021, 10(11): 1746.
[85] Chouchani ET, Kazak L, Spiegelman BM. New advances in adaptive thermogenesis: UCP1 and beyond., 2019, 29(1): 27–37.
[86] Mills EL, Harmon C, Jedrychowski MP, Xiao HP, Garrity R, Tran NV, Bradshaw GA, Fu A, Szpyt J, Reddy A, Prendeville H, Danial NN, Gygi SP, Lynch L, Chouchani ET. UCP1 governs liver extracellular succinate and inflammatory pathogenesis., 2021, 3(5): 604–617.
[87] De Munck TJI, Xu P, Vanderfeesten BLJ, Elizalde M, Masclee AAM, Nevens F, Cassiman D, Schaap FG, Jonkers DMAE, Verbeek J. The role of brown adipose tissue in the development and treatment of nonalcoholic steatohepatitis: an exploratory gene expression study in mice., 2020, 52(12): 869–876.
[88] Gallardo-Montejano VI, Yang CF, Hahner L, McAfee JL, Johnson JA, Holland WL, Fernandez-Valdivia R, Bickel PE. Perilipin 5 links mitochondrial uncoupled respiration in brown fat to healthy white fat remodeling and systemic glucose tolerance., 2021, 12(1): 3320.
[89] Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection., 2018, 1859(9): 940–950.
[90] Seshadri N, Jonasson ME, Hunt KL, Xiang B, Cooper S, Wheeler MB, Dolinsky VW, Doucette CA. Uncoupling protein 2 regulates daily rhythms of insulin secretion capacity in MIN6 cells and isolated islets from male mice., 2017, 6(7): 760–769.
[91] Li JR, Jiang RH, Cong XL, Zhao YF. UCP2 gene polymorphisms in obesity and diabetes, and the role of UCP2 in cancer., 2019, 593(18): 2525–2534.
[92] Camara Y, Mampel T, Armengol J, Villarroya F, Dejean L. UCP3 expression in liver modulates gene expression and oxidative metabolism in response to fatty acids, and sensitizes mitochondria to permeability transition., 2009, 24(3-4): 243–252.
[93] Tan MJ, Mosaoa R, Graham GT, Kasprzyk-Pawelec A, Gadre S, Parasido E, Catalina-Rodriguez O, Foley P, Giaccone G, Cheema A, Kallakury B, Albanese C, Yi CL, Avantaggiati ML. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/ NASH., 2020, 27(7): 2143–2157.
[94] Unami A, Shinohara Y, Kajimoto K, Baba Y. Comparison of gene expression profiles between white and brown adipose tissues of rat by microarray analysis., 2004, 67(3): 555–564.
[95] Mizuarai S, Miki S, Araki H, Takahashi K, Kotani H. Identification of dicarboxylate carrier Slc25a10 as malate transporter in de novo fatty acid synthesis., 2005, 280(37): 32434–32441.
[96] Lin Y, Berg AH, Iyengar P, Lam TKT, Giacca A, Combs TP, Rajala MW, Du XL, Rollman B, Li WJ, Hawkins M, Barzilai N, Rhodes CJ, Fantus IG, Brownlee M, Scherer PE. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species., 2005, 280(6): 4617–4626.
[97] An YA, Chen S, Deng YF, Wang ZV, Funcke JB, Shah M, Shan B, Gordillo R, Yoshino J, Klein S, Kusminski CM, Scherer PE. The mitochondrial dicarboxylate carrier prevents hepatic lipotoxicity by inhibiting white adipocyte lipolysis., 2021, 75(2): 387–399.
[98] Zhong Q, Putt DA, Xu F, Lash LH. Hepatic mitochondrial transport of glutathione: studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells., 2008, 474(1): 119–127.
[99] Pan GQ, Wang RB, Jia SN, Li YQ, Jiao Y, Liu N. SLC25A11 serves as a novel prognostic biomarker in liver cancer., 2020, 10(1): 9871.
[100] Yuan YX, Zhu CJ, Wang YL, Sun J, Feng JL, Ma ZW, Li PL, Peng WT, Yin C, Xu GL, Xu PW, Jiang YW, Jiang QY, Shu G. α-Ketoglutaric acid ameliorates hyperglycemia in diabetes by inhibiting hepatic gluconeogenesis via serpina1e signaling., 2022, 8(18): eabn2879.
[101] Palmieri F, Pierri CL. Mitochondrial metabolite transport., 2010, 47: 37–52.
[102] Saheki T, Kobayashi K, Iijima M, Moriyama M, Yazaki M, Takei YI, Ikeda SI. Metabolic derangements in deficiency of citrin, a liver-type mitochondrial aspartate- glutamate carrier., 2005, 33(2): 181–184.
[103] Amoedo ND, Punzi G, Obre E, Lacombe D, De Grassi A, Pierri CL, Rossignol R. AGC1/2, the mitochondrial aspartate-glutamate carriers., 2016, 1863(10): 2394–2412.
[104] Komatsu M, Yazaki M, Tanaka N, Sano K, Hashimoto E, Takei YI, Song YZ, Tanaka E, Kiyosawa K, Saheki T, Aoyama T, Kobayashi K. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease., 2008, 49(5): 810–820.
[105] Tonazzi A, Giangregorio N, Console L, Palmieri F, Indiveri C. The mitochondrial carnitine acyl-carnitine carrier (SLC25A20): molecular mechanisms of transport, role in redox sensing and interaction with drugs., 2021, 11(4): 521.
[106] Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution., 2004, 279(29): 30722–30730.
[107] Harborne SPD, King MS, Crichton PG, Kunji ERS. Calcium regulation of the human mitochondrial ATP- Mg/Pi carrier SLC25A24 uses a locking pin mechanism., 2017, 7: 45383.
[108] Urano T, Shiraki M, Sasaki N, Ouchi Y, Inoue S. SLC25A24 as a novel susceptibility gene for low fat mass in humans and mice., 2015, 100(4): E655–E663.
[109] Black PN, DiRusso CC. Vectorial acylation: linking fatty acid transport and activation to metabolic trafficking., 2007, 286: 127–138.
[110] Stahl A, Evans JG, Pattel S, Hirsch D, Lodish HF. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes., 2002, 2(4): 477–488.
[111] Krammer J, Digel M, Ehehalt F, Stremmel W, Füllekrug J, Ehehalt R. Overexpression of CD36 and acyl-CoA synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in human hepatoma cells., 2011, 8(7): 599–614.
[112] Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, Stahl A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase., 2010, 299(3): E384– E393.
[113] Doege H, Baillie RA, Ortegon AM, Tsang B, Wu QW, Punreddy S, Hirsch D, Watson N, Gimeno RE, Stahl A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis., 2006, 130(4): 1245–1258.
[114] Hubbard B, Doege H, Punreddy S, Wu H, Huang XM, Kaushik VK, Mozell RL, Byrnes JJ, Stricker-Krongrad A, Chou CJ, Tartaglia LA, Lodish HF, Stahl A, Gimeno RE. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity., 2006, 130(4): 1259–1269.
[115] Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, Shimizu T, Mita T, Kanazawa A, Imaizumi MO, Abe T, Kiyonari H, Hojyo S, Fukada T, Kawauchi T, Nagamatsu S, Hirano T, Kawamori R, Watada H. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance., 2013, 123(10): 4513–4524.
[116] Hardy AB, Wijesekara N, Genkin I, Prentice KJ, Bhattacharjee A, Kong D, Chimienti F, Wheeler MB. Effects of high-fat diet feeding on Znt8-null mice: differences between β-cell and global knockout of Znt8., 2012, 302(9): E1084– E1096.
[117] Fukunaka A, Fujitani Y. Role of zinc homeostasis in the pathogenesis of diabetes and obesity., 2018, 19(2): 476.
[118] Mattace Raso G, Simeoli R, Russo R, Iacono A, Santoro A, Paciello O, Ferrante MC, Canani RB, Calignano A, Meli R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet., 2013, 8(7): e68626.
[119] Li XL, Li J, Lu XL, Ma HH, Shi HT, Li H, Xie DH, Dong L, Liang CL. Treatment with PPARδ agonist alleviates non-alcoholic fatty liver disease by modulating glucose and fatty acid metabolic enzymes in a rat model., 2015, 36(3): 767–775.
[120] Wikan N, Tocharus J, Sivasinprasasn S, Kongkaew A, Chaichompoo W, Suksamrarn A, Tocharus C. Capsaicinoid nonivamide improves nonalcoholic fatty liver disease in rats fed a high-fat diet., 2020, 143(3): 188–198.
[121] Zhang Y, Lu QH, Cao HF, Zhang SR, Wang HD. Effects of GPR81 agonist on insulin resistance in rats with nonalcoholic fatty liver disease., 2021, 37(4): 354–358.
[122] Lin W, Jin Y, Hu X, Huang EJ, Zhu QH. AMPK/PGC- 1α/GLUT4-mediated effect of icariin on hyperlipidemia- induced non-alcoholic fatty liver disease and lipid metabolism disorder in mice., 2021, 86(11): 1407–1417.
[123] Yasmin T, Rahman MM, Khan F, Kabir F, Nahar K, Lasker S, Islam MD, Hossain MM, Hasan R, Rana S, Alam MA. Metformin treatment reverses high fat diet- induced non-alcoholic fatty liver diseases and dyslipidemia by stimulating multiple antioxidant and anti-inflammatory pathways., 2021, 28: 101168.
[124] Ma L, Lian YL, Tang JY, Chen FY, Gao H, Zhou Z, Hou N, Yi W. Identification of the anti-fungal drug fenticonazole nitrate as a novel PPARγ-modulating ligand with good therapeutic index: structure-based screening and biological validation., 2021, 173: 105860.
[125] Zaklos-Szyda M, Pietrzyk N, Kowalska-Baron A, Nowak A, Chałaśkiewicz K, Ratajewski M, Budryn G, Koziołkiewicz M. Phenolics-rich extracts of dietary plants as regulators of fructose uptake in Caco-2 cells via GLUT5 involvement., 2021, 26(16): 4745.
[126] Nasykhova YA, Tonyan ZN, Mikhailova AA, Danilova MM, Glotov AS. Pharmacogenetics of type 2 diabetes- progress and prospects., 2020, 21(18): 6842.
[127] Huard K, Brown J, Jones JC, Cabral S, Futatsugi K, Gorgoglione M, Lanba A, Vera NB, Zhu YM, Yan QY, Zhou YJ, Vernochet C, Riccardi K, Wolford A, Pirman D, Niosi M, Aspnes G, Herr M, Genung NE, Magee TV, Uccello DP, Loria P, Di L, Gosset JR, Hepworth D, Rolph T, Pfefferkorn JA, Erion DM. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5)., 2015, 5: 17391.
[128] Sun QS, Niu Q, Guo YT, Zhuang Y, Li XN, Liu J, Li N, Li ZY, Huang F, Qiu ZX. Regulation on citrate influx and metabolism through inhibiting SLC13A5 and ACLY: a novel mechanism mediating the therapeutic effects of curcumin on NAFLD., 2021, 69(31): 8714–8725.
[129] Savic D, Hodson L, Neubauer S, Pavlides M. The importance of the fatty acid transporter L-carnitine in non-alcoholic fatty liver disease (NAFLD)., 2020, 12(8): 2178.
[130] Weng Y, Fonseca KR, Bi YA, Mathialagan S, Riccardi K, Tseng E, Bessire AJ, Cerny MA, Tess DA, Rodrigues AD, Kalgutkar AS, Litchfield JE, Di L, Varma MVS. Transporter-enzyme interplay in the pharmacokinetics of PF-06835919, a first-in-class ketohexokinase inhibitor for metabolic disorders and non-alcoholic fatty liver disease., 2022, 50(9): 1312–1321.
[131] Zhao ST, Chu Y, Zhang YW, Zhou YL, Jiang ZW, Wang ZQ, Mao LF, Li K, Sun W, Li P, Jia SQ, Wang CC, Xu AM, Loomes K, Tang SB, Wu DH, Hui XY, Nie T. Linifanib exerts dual anti-obesity effect by regulating adipocyte browning and formation., 2019, 222: 117–124.
[132] Colca JR, McDonald WG, Adams WJ. MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis., 2018, 27(7): 631–636.
[133] Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, Kowdley KV, Lai M, Schiff E, Parmar D, Patel P, Chalasani N. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double- blind phase 2 trial., 2021, 74(4): 1809–1824.
[134] Sven MF, Pierre B, Manal FA, Quentin MA, Elisabetta B, Vlad R, Philippe HM, Bruno S, Jean-Louis J, Pierre B, Jean-Louis A. A randomised, double-blind, placebo- controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study., 2020, 98: 106170.
[135] Bhattacharya D, Basta B, Mato JM, Craig A, Fernández-Ramos D, Lopitz-Otsoa F, Tsvirkun D, Hayardeny L, Chandar V, Schwartz RE, Villanueva A, Friedman SL. Aramchol downregulates stearoyl CoA- desaturase 1 in hepatic stellate cells to attenuate cellular fibrogenesis., 2021, 3(3): 100237.
[136] Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, Arrese M, Fracanzani AL, Ben Bashat D, Lackner K, Gorfine T, Kadosh S, Oren R, Halperin M, Hayardeny L, Loomba R, Friedman S, ARREST Investigator Study Group, Sanyal AJ. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial., 2021, 27(10): 1825–1835.
Progress of solute carrier SLC family in nonalcoholic fatty liver disease
Zhiquan Tang, Li Shi, Jing Xiong
Nonalcoholic fatty liver disease is closely related to obesity and type 2 diabetes mellitus, and is one of the components of metabolic syndrome. Due to the complexity of its pathogenesis, there is no effective drug treatment to date. Solute carrier transporters are associated with a variety of metabolic diseases and are abundantly expressed in the liver. They participate in the transport of a variety of nutrients and metabolites, regulate nutrient supply, metabolic transformation, energy balance and oxidative stress, and modulate the physiological functions of liver. Particularly, it is important that some of these SLC transporters have become new targets for drug development. In this review, we summarize the role of SLC in the transport of nutrients and liver metabolites and its correlation with NAFLD, and reveal the potential of SLC as a target for the development of new drugs for NAFLD treatment so as to provide a new choice for the treatment of the disease.
nonalcoholic fatty liver disease; solute carrier SLC family; glucose and lipid metabolism
2022-07-16;
2022-09-22;
2022-09-30
国家自然科学基金项目(编号:82070883,82273982),江苏省自然科学基金项目(编号:BK20221525)和中国药科大学高层次人才科研启动项目资助[Supported by the National Natural Science Foundation of China (Nos. 82070883, 82273982), the National Natural Science Foundation of Jiangsu Province (No. BK20221525), and the Scientific Research Foundation for High-level Faculty, China Pharmaceutical University]
汤志全,在读硕士研究生,专业方向:药理学。E-mail: 1350312679@qq.com
熊晶,博士,研究员,研究方向:靶向代谢稳态研究肝脏疾病的分子机制与药物干预。E-mail: jxiong@cpu.edu.cn
10.16288/j.yczz.22-238
(责任编委: 孟卓贤)
