石墨烯基氮掺杂层级孔碳负载钯及其PTA加氢精制应用
2022-11-18张晓利杨叶伟何观伟
殷 迪,郑 晴,张晓利,卞 雯,杨叶伟,何观伟
(西北化工研究院有限公司,陕西 西安 710061)
精对苯二甲酸(PTA)是一种重要的有机二元酸,广泛用于聚酯纤维、聚酯瓶片和聚酯薄膜的生产。PTA工业化生产的典型工艺为两步法精PTA工艺,首先以对二甲苯(PX)为原料,在催化剂作用下氧化制得粗对苯二甲酸。粗对苯二甲酸中含有的副产物4-羧基苯甲醛(4-CBA),会影响PTA产品质量以及后续聚酯的加工性能,需要采用加氢精制步骤除去。PTA的加氢精制反应是指在钯炭催化剂作用下,在温度(270~290) ℃、压力(7.0~8.0) MPa条件下,将粗对苯二甲酸中的4-CBA还原成易溶于水的对甲基苯甲酸,再经结晶、分离、干燥制得纤维级PTA[1-3]。
钯是粗对苯二甲酸加氢精制使用最广泛的活性组分,工业化生产中常用的催化剂为活性炭负载的钯催化剂,但仍存在一些挑战。一方面,活性炭因其中巨大的微孔结构不利于分子的扩散,必须精心设计碳材料的多孔结构以强化传质从而提高催化效率;另一方面,由于活性炭其表面化学惰性,与金属的相互作用较弱,活性金属物种在活性炭上表现出较大的聚集和浸出倾向,导致活性金属粒子的流失和团聚。因此,研究者尝试对载体进行修饰,以增强这种相互作用。
层级孔碳材料是一类新型的多孔碳材料[4-8],同时兼具不同尺寸与功能的微孔、中孔或大孔。层级孔碳材料是较理想的金属催化剂载体,其大孔可以阻碍所负载颗粒的团聚,介孔提供了反应溶液的输运通道,微孔促进了生成气体的扩散,大比表面积有助于催化活性位和反应物的接触。一般来说,纯碳材料的亲水性较差,而这在一定程度上掣肘了其在各个方面的应用。在元素周期表中氮原子位于碳原子相邻位置,因而可取代碳材料骨架中的碳原子,掺杂在碳材料中的氮原子可以在材料表面结构的改变、孔道结构的调节、亲水性的增强、对材料表面pKa值的影响、材料电子传输速率的改善等方面起到极大作用,从而拓展碳材料的应用范围[9-11]。碳材料中掺杂的N原子可有效地提高催化剂的催化活性以及催化剂的寿命,显著改善其吸附及催化性能;有利于降低贵金属的用量,减少催化剂制备成本;提高碳材料对氢气的吸附能力。
本文以氧化石墨烯为结构导向剂,间苯二酚-甲醛-三聚氰胺为前驱体,其中三聚氰胺为氮源,经溶胶-凝胶法及炭化制备得到三维堆叠石墨烯基氮掺杂层级孔碳纳米片(NCNS),负载Pd后制得的Pd/NCNS催化剂用于PTA加氢精制,探索NCNS孔道结构及氮含量的调控机制,重点考察负载Pd分散度与氮含量的相关性以及载体孔道结构及氮含量对催化活性的影响。
1 实验部分
1.1 原料
氧化石墨烯(GO),12 mg·mL-1,中国科学院山西煤炭化学研究所;甲醛(37%)、苯二酚、三聚氰胺、氯钯酸钠、盐酸(37%)、氢氧化钠、甲酸钠,分析纯;Pd/C催化剂,0.5%;粗对苯二甲酸,含4-CBA质量分数3%,中国石化上海石油化工研究院。
1.2 催化剂制备
1.2.1 碳纳米片制备
将一定质量的间苯二酚(R)与甲醛溶液(F,质量分数37%)混合(R与 F的物质的量比为1∶2),在40 ℃的恒温水浴中反应30 min,得到RF溶液。将三聚氰胺(M)在80 ℃溶解于甲醛溶液(F)中搅拌15 min(M与F的物质的量比为 1∶3),得到MF溶液。将RF和MF溶液混合,即得到一定浓度的RMF溶液,加入一定量经过超声处理的氧化石墨烯水分散液(RMF与GO 的质量比分别为 40、60、80、100),将所得的混合液在80 ℃水浴中搅拌24 h,抽滤、80 ℃干燥,得到聚合物RMF-GO粉末。
将所得的聚合物氧化石墨烯纳米片在N2气流保护下,以1 ℃·min-1的升温速率升至800 ℃,并停留3 h,即可得到含氮多孔炭-氧化石墨烯纳米片材料,命名为NCNS-x(y),其中x为M与R物质的量比,y为RMF和氧化石墨烯质量比。
1.2.2 Pd的负载
称取含氯化钯4.2 g的氯钯酸钠溶液23.1 g,加入200 g去离子水配制成浸渍胶液,调节浸渍胶液pH=4.2,5 min内将溶液倒在500 g碳载体上,并于室温静置12 h。浸渍后的催化剂前驱体在空气中老化6 h。取200 g浓度为1%的甲酸钠溶液配制成水溶液,水溶液的量以刚好浸没所有催化剂前驱体为宜,在120 ℃还原120 min,将负载于碳载体上的Pd化合物还原成金属Pd,然后用去离子水洗涤,至溶液呈中性或无氯离子为止。过滤后即得Pd负载质量分数为0.5%的Pd/NCNS催化剂。
1.3 催化剂表征
孔道结构在美国康塔公司(Quadrasorb SI)全自动氮气吸附仪上于77 K条件下进行测定,采用BET方法计算样品的比表面积,QS-DFT方法计算孔径分布,在吸附量最大值处计算总孔容,采用T-plot法计算微孔表面积及微孔孔容。
采用日本电子株式会社7100F型场发射扫描电子显微镜(SEM)观察样品的表面形貌。
样品中的C、N、H元素含量由德国Elementar Vario ELⅢ 元素分析仪测得,O元素的含量则采用差量法计算。
X射线衍射图谱由日本理学公司 D/max 2550型X射线衍射仪测定,CuKα,λ=0.154 06 nm,工作电流为40 mA,工作电压为40 kV,以0.010°·s-1速率进行扫描。
样品的表面组成采用瑞典VG Scientific公司的ESCALab220i-XL型光电子能谱仪测定,激发源为单色化A1K X射线,功率为300 W,分析时的基础真空为3×10-7Pa。
采用H2-O2滴定法测定样品的Pd分散度、活性表面积和晶粒度,在美国麦克仪器公司AutoChemⅡ2920化学吸附仪上进行。样品首先在氢气气氛中200 ℃还原处理,再切换惰气气氛吹扫至基线平稳,切换氧气进行氧化,然后用脉冲氢气还原氧化物,根据耗氢量即可计算表面金属钯的原子数,进一步计算得到Pd的分散度、活性表面积和晶粒度。
1.4 催化剂活性评价
催化剂活性评价在不锈钢搅拌间歇高压反应釜中进行,评价条件为:催化剂装填量为2.0 g,粗对苯二甲酸30.0 g,含4-CBA为1.0 g,水溶液900.0 mL,反应压力7.5 MPa,反应温度280 ℃。反应后液体产物通过高效液相色谱配紫外检测器进行定量分析,通过计算剩余的4-CBA含量评价催化剂活性,剩余的4-CBA含量越低,说明催化剂的催化加氢效率越高。
2 结果与讨论
2.1 碳载体制备
采用溶胶-凝胶法结合结构导向支撑骨架制备氮掺杂碳纳米片,关键点在于如何在二维结构导向剂氧化石墨烯(GO)表面均匀地包覆树脂聚合物,从而形成典型的三明治多层结构。因为氧化石墨烯表面具有羟基等大量的亲水性官能团,GO可以稳定地分散于水中,形成均匀的水分散液。而间苯二酚-甲醛-三聚氰胺(RMF)水溶液其表面则呈现出一定的正电荷性质,当二者混合时,可以通过静电引力作用而使得RMF酚醛树脂在GO表面进行快速聚合反应,进而形成层状包覆结构。通过调节GO和RMF酚醛树脂的质量比,即可制得具有不同包覆厚度的纳米片层结构。而通过调节前驱体中三聚氰胺和间苯二酚物质的量比,可以控制碳纳米片的氮含量。
2.1.1 RMF与GO质量比
通过改变结构导向剂GO和包覆聚合物的质量比,制备得到具有不同包覆厚度的纳米片层结构。图1为不同RMF与GO质量比条件下得到的样品炭化后的扫描电镜照片。

图1 不同RMF与GO质量比的氮掺杂碳纳米片的扫描电镜照片Figure 1 SEM images of nitrogen doped carbon nanosheets with various mass ratio of RMF/GO
由图1可以明显看出,随着RMF与GO质量比增大,纳米片层结构的厚度依次增加,当RMF与GO质量比为40时,片层厚度约为20 nm;当RMF与GO质量比为60时,片层厚度约增加到50 nm;而当RMF与GO质量比提高到80和100时,片层厚度约为60 nm和90 nm。因而,通过改变聚合物添加量,可以有效地对纳米片层厚度进行调节。值得注意的是,整个体系中RMF树脂在氧化石墨烯表面包覆均匀,没有自我聚合而形成聚合物微球,表明GO对其起到了优异的结构导向作用。
图2为不同RMF与GO质量比得到的碳纳米片的氮气吸附-脱附等温线和DFT孔径分布。从图2可以看到,所有的样品在较低的相对压力下,均具有较大的氮气吸附量,表明材料中存在一定数量的微孔,同时随着聚合物用量的增大,微孔比例也逐渐增大(表1)。但当聚合物用量较低时,脱附曲线具有典型的H2型滞后环,表现出一定程度的类石墨烯的结构,即狭窄的片层堆积而成的孔道结构。随着RMF聚合物用量增加,纳米片层厚度不断增大,更明显地表现为纯微孔结构,其H2滞回环也逐渐消失。表明纳米片层最终都表现为RMF聚合物的分子链接形成的孔道,氧化石墨烯对孔结构的贡献则越来越小。

图2 不同RMF与GO质量比氮掺杂碳纳米片的氮气吸附-脱附等温线和孔径分布Figure 2 N2 adsorption-desorption isotherms,and DFT pore size distribution of nitrogen doped carbon nanosheets with various mass ratio of RMF/GO
样品孔结构参数如表1所示。由表1可以看出,随着RMF与GO质量比增大,样品的比表面积均在约1 000 m2·g-1波动,微孔比表面积则在约950 m2·g-1波动,两者的变化均不明显。采用差减法可知中孔孔容则从0.32 cm3·g-1逐渐减小到0.16 cm3·g-1、0.12 cm3·g-1和0.11 cm3·g-1,正好对应图2脱附曲线H2滞回环面积的逐渐减小。

表1 不同RMF与GO质量比氮掺杂碳纳米片的孔结构参数
表2是不同RMF与GO质量比氮掺杂碳纳米片的CNH元素分析结果。从表2可见,随RMF与GO质量比增加,氮含量从NCNS-1(40)的9.14%缓慢增加到NCNS-1(100)的10.52%,碳元素的含量呈现缓慢增加趋势,氢元素基本持平,而氧元素则从NCNS-1(40)的8.56%逐渐减小到NCNS-1(100)的4.73%。这是因为前驱体RMF-GO中GO的含氧量比RMF高,因此,前驱体中GO比例越小,碳化后碳纳米片的含氧量也越小;反之,氮元素几乎全部来自RMF中的三聚氰胺,随着前驱体中RMF比例增大,碳化后碳纳米片的氮含量也逐渐增大,碳元素含量随之提高。但由于RMF中M与R的比例维持在1∶1,因此样品的氮碳比接近0.13。

表2 不同RMF与GO质量比氮掺杂碳纳米片的元素组成
2.1.2 三聚氰胺含量
图3为不同M与R物质的量比的氮掺杂碳纳米片的扫描电镜照片。从图3可以看出,三个样品均呈现相类似的片层结构,片层厚度约20 nm,表明M与R物质的量比对氮掺杂碳纳米片的形貌没有明显影响。

图3 不同M与R物质的量比氮掺杂碳纳米片的扫描电镜照片Figure 3 SEM images of nitrogen doped carbon nanosheets with various mole ratio of M/R
图4为不同M与R物质的量比得到的氮掺杂碳纳米片的氮气吸附-脱附等温线和DFT孔径分布。由图4可见,所有样品的氮气吸附-脱附曲线在相对压力较低时呈现快速上升趋势,接着吸附曲线变得相对平坦,表现出Ⅰ型吸附等温线特征(按IUPAC分类),说明样品呈现出典型的微孔结构。同时可以观察到,在M与R物质的量比为0.5、1和2时,样品在低相对压力处的吸附量类似,这反映在样品的微孔比表面积从M与R物质的量比为0.5时的865 m2·g-1增加到M与R物质的量比为1时的932 m2·g-1和M与R物质的量比为2时的1 003 m2·g-1(表3),因前驱体中氮含量愈高,聚合物分解速率越快,反应越剧烈,随着分解程度的提高产生出更多微孔。M与R物质的量比为0.5和1的样品存在明显的H2型滞回环,M与R物质的量比为0.5的面积更大,说明在这两个样品中存在典型的类石墨烯结构,而M与R物质的量比为2的样品则无明显的H2滞回环,呈现单纯微孔结构,采用差减法得三个样品中孔孔容分别为0.72 cm3·g-1、0.32 cm3·g-1、0.07 cm3·g-1,呈逐渐下降趋势,这是因为,随着分解剧烈程度的提高,片层之间黏连堆叠的程度也相应提高,导致片层间中大孔减少。

图4 不同M与R物质的量比氮掺杂碳纳米片的氮气吸附-脱附等温线和孔径分布Figure 4 N2 adsorption-desorption isotherms and DFT pore size distribution of nitrogen doped carbon nanosheets with various mole ratio of M/R
表4为不同M与R物质的量比氮掺杂碳纳米片的CNH元素分析结果。从表4可见,随着M与R物质的量比增加,氮含量呈快速增加趋势,M与R物质的量比为0.5、1、2时的氮含量分别为6.38%、9.14%和13.49%。碳含量则呈减少趋势,从82.82%逐渐减少到73.19%。氮碳比从0.09增加到0.20,也增加两倍。说明通过调控M与R物质的量比,可以实现掺氮量可控的碳纳米片制备。

表3 不同M与R物质的量比氮掺杂碳纳米片的孔结构参数
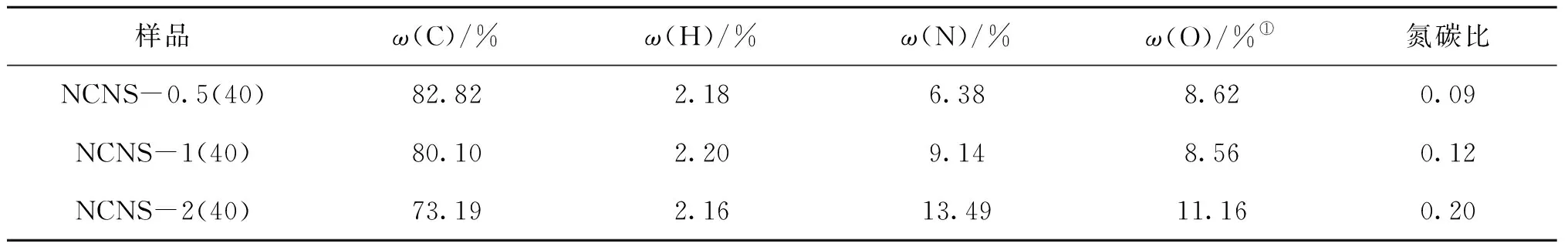
表4 不同M与R物质的量比氮掺杂碳纳米片的元素组成
图5为不同M与R物质的量比氮掺杂碳纳米片的XPS分析结果。由图5可知,XPS全谱扫描曲线中出现明显的N1s信号峰,说明N元素已被成功掺杂到碳纳米片的碳骨架结构中。与此同时,随着M与R物质的量比增加,样品的N1s信号峰强度逐渐增强,表明碳纳米片中掺杂的N元素含量可以通过改变M与R物质的量比调控。由图5还可以看出,所有氮掺杂碳纳米片的N1s峰均可在结合能为398.6 eV、399.9 eV和400.9 eV处分为三个峰,分别对应于吡啶氮(N-6)、吡咯氮(N-5)和石墨化氮(N-Q)。吡啶氮(N-6)以六元环的结构形式存在于碳层石墨环的边缘,并与两个碳原子相连,在其外层电子轨道存在一对未成键孤对电子,因而呈现出强的碱性。吡咯氮(N-5)则以五元环结构形式存在于碳层石墨环的边缘,其为石墨环的离域π系统供应了一对P电子,从而强化了碳材料对小分子气体的界面吸附能力。石墨化氮(N-Q)位于石墨烯片层的中心位置,其以sp2杂化轨道结构形式与三个碳原子相连,同时参与了石墨六元环中离域大π键的形成,从而强化了石墨烯片层表面的碱性。

图5 不同M与R物质的量比氮掺杂碳纳米片的XPS全谱扫描曲线、XPS N1s及其分峰结果、不同氮种类示意图Figure 5 XPS survey,XPS N1s and peak division,and schematic diagram for different nitrogen species of NCNSs with various mole ratio of M/R
2.2 Pd负载量
图6给出氮掺杂碳纳米片负载钯的Pd/NCNS及商用Pd/C的XRD图。

图6 氮掺杂碳纳米片负载钯及商用Pd/C的XRD图Figure 6 XRD patterns of Pd/NCNSs and commercial Pd/C
由图6可以看出,所有样品的出峰位置以及峰形基本保持一致,其在2θ=23.7°以及43.6°两处均呈现出异常弥散的衍射峰,这两个峰分别对应于石墨微晶结构的[002]和[100]晶面,表明样品均呈现出典型的无定型炭结构。值得注意的是,随着M与R物质的量比即氮含量的增加,样品对应于石墨[002]晶面的峰形渐渐变宽,同时峰位置向左轻微逐渐偏移,说明样品的石墨化度呈现降低的趋势,而且类石墨微晶层间距逐渐轻微变宽。在XRD图中没有观察到归属Pd晶体的衍射峰,说明此时Pd纳米颗粒被高度分散在载体表面,且其晶粒尺寸非常小,超出XRD仪器的检测限。
因为XRD图谱没有给出Pd晶粒的特征峰,为了表征Pd晶粒分散度、活性比表面积和晶粒度,采用H2-O2滴定法进行了上述工作,结果示于表5。由表5可知,氮掺杂碳纳米片负载Pd的分散度要显著高于商用Pd/C,具有较大活性比表面积和较小晶粒度;值得注意的是,随着氮含量的提高,Pd分散度也明显上升。这些结果说明氮掺杂有助于Pd在碳载体表面的分散。由表5还可以看出,负载Pd后,所有样品的比表面积都有小幅度的降低,平均孔径为(0.61~0.67) nm,这是N2吸附的检测下限,而商用Pd/C的平均孔径却为2.5 nm,这是因为碳纳米片只经过碳化,而商用Pd/C的活性炭载体经过了活化步骤。由于选择DFT计算方法,碳纳米片堆叠形成的介孔及大孔无法在此处体现。

表5 氮掺杂碳纳米片负载钯及商用Pd/C的H2-O2滴定结果及孔结构参数
2.3 活性评价结果
样品的活性评价数据及相关联的表征参数列于表6。由表6可知,4-CBA转化率随样品氮含量及Pd分散度的增加而提高。商用Pd/C催化剂不含氮,Pd分散度仅为23.9%,4-CBA转化率为96.3%;而氮含量最高的Pd/NCNS-2(40)催化剂,其Pd分散度也是样品中最高的(55.1%),4-CBA转化率达99.6%。这是因为氮掺杂所致的Pd分散度愈高,暴露在表面的活性位愈多,同时氮还可能起到了吸附活化氢气和4-CBA的作用。此外,对于含氮量类似,但片层厚度不同的Pd/NCNS-1系列样品,随着片层厚度增加,其活性呈缓慢下降趋势,这可能是扩散的影响。氮掺杂碳纳米片负载钯催化剂活性高于商用Pd/C催化剂,其扩散通道较短也是重要的影响因素,活性炭的孔道由错综复杂的微孔构成,其长度远大于碳纳米片的厚度,而碳纳米片堆叠而成的三维结构,片层间的中大孔起到了主要的传质作用。

表6 氮掺杂碳纳米片负载钯催化剂及商用Pd/C催化剂的活性及相关表征参数
3 结 论
(1) 通过调节原料中间苯二酚-甲醛-三聚氰胺与氧化石墨烯的比例,经过溶胶-凝胶法及炭化可以制备出厚度均一可控的氮掺杂碳纳米片材料,当RMF与GO的质量比分别为40、60、80、100时,碳化后样品的片层厚度分别为20 nm、50 nm、60 nm、90 nm。通过控制三聚氰胺与间苯二酚物质的量比,可以调节碳纳米片的氮含量。当M与R物质的量比分别为0.5、1和2时,所得碳纳米片的氮含量分别为6.38%、9.14%、13.49%。
(2) 氮掺杂可提高碳载体上Pd分散度,不含氮的商用Pd/C催化剂上Pd分散度仅为23.9%,氮含量最高的Pd/NCNS-2(40)催化剂上Pd分散度可达55.1%。
(3) 氮掺杂碳纳米片负载钯催化剂表现出比商用Pd/C催化剂更高的活性,归因于其较短的扩散通道、氮掺杂及其所致的高Pd分散度。
