MnZSM-5 催化剂上NH3-SCR 反应机理的研究
2022-11-15李宝忠王宽岭李英霞
李宝忠, 王宽岭, 李英霞
(1. 中国石油化工股份有限公司 大连石油化工研究院, 辽宁 大连 116045;2. 北京化工大学 化工资源有效利用国家重点实验室, 北京 100029)
化石燃料的燃烧过程中会产生大量的氮氧化物(NOx), 特别是工业生产中窑炉的燃烧和机动车尾气排放, 这对人体和环境造成了严重的影响, 因此NOx排放控制的重要性不容忽视[1-2]. NOx主要包括NO、 NO2、 N2O、 N2O5等, 其中NO为主要成分, 约占90%~95%, 目前NH3选择性催化还原(NH3-SCR) 是最有效且应用最广的NOx控制方法[3-5]. NH3-SCR技术的核心就是脱硝催化剂, 锐钛矿TiO2负载氧化钒是NH3-SCR反应的传统催化剂, 但由于氧化钒具有毒性, 反应条件要求较高, 应用受到限制. 因此, 开发绿色环保、 高效无毒、 来源广泛的新型催化剂是脱硝反应的技术关键, 其中, 锐钛矿TiO2负载型过渡金属和稀土金属基低温脱硝催化剂是研究的热点, 理论和应用研究取得了重大进展[6-7].
近些年, 分子筛负载金属氧化物催化剂显示出高催化活性和N2选择性, 作为NH3-SCR反应催化剂的潜力股受到广泛关注[8-10]. Mn因半充满状态的电子排布具有较强的氧化还原能力, 负载型锰基催化剂在NH3-SCR反应中更是体现出优异的催化能力.较多研究者对分子筛负载锰基催化剂在NH3-SCR反应中的应用进行了大量实验研究[11-13], 如, Lou等[11]研究了焙烧温度对采用沉淀法制备出的Mn/ZSM-5催化剂表面活性物种的影响, 结果表明300 ℃时可得到活性最高的Mn/ZSM-5, 该温度下Mn主要以Mn3O4和非晶型MnO2形态存在于催化剂表面,其在表面的富集使得催化剂活性优越. 研究证明Mn基催化剂具有良好的催化活性和催化选择性, 但在反应机理研究方面需要进一步研究, NO和NH3在催化剂上吸附状态、 形成的重要中间体、 遵循的反应机理需要明晰, 反应过程中的中间产物活化转化过程仍需探究. 我们将从分子层面上对Mn/ZSM-5催化剂上NH3-SCR反应机理进行理论研究.
NH3-SCR反应是指NOx在还原剂作用下反应生成无污染的N2和H2O, 反应方程式见(1)、 (2):

Mao等[14]对Cu-SAPO-34 催化剂上NH3-SCR反应机理进行了理论研究, 认为该反应是一个氧化还原过程, 伴随着铜离子的氧化还原, NOx被还原为N2和H2O. 经大量实验和理论研究工作, 目前得到广泛认可的机理分为两种, Eley-Rideal(E-R)机理和Langmuir-Hinshelwood(L-H)机理[15-16], E-R机理为气态NO直接加入反应, 而L-H机理为气态NO先在活性位上吸附活化, 而后再在NH3-SCR反应中发挥作用. E-R机理和L-H机理的本质差异在于NO分子参加反应的形式, 如气态、 吸附态等.
Yu等[17]通过实验的方式对分子筛SAPO-34负载MnOx催化剂上NH3-SCR反应机理进行了系列研究, 认为NH3-SCR反应中还原剂NH3吸附活化后形成的[NH2]是反应的关键, 气相NO与吸附态[NO]均能与之反应, 因此认为可能存在遵循E-R机理或L-H机理的两条反应路径. Chen等[18]对采用凝胶法制备的Mn/TiWOx和Mn/TiO2催化剂上NH3-SCR反应进行研究时, 发现吸附态的NH3比吸附态的NOx消耗得快得多, 推断吸附态的NH3更易与气态NOx反应, 即反应遵循E-R机理.
因实验存在一定局限性, 无法从分子层面剖析反应机理, 因此可在实验的基础上, 从微观层面对NH3-SCR反应可能存在的反应机理进行理论计算,加深研究者对其理解和掌握, 从而更有针对性和目的性地开发无毒高效的新型催化剂.
1 实验部分
所有计算均利用高斯09程序[19]中密度泛函理论(DFT)B3LYP进行, 活性组分Mn使用SDD基组,其余原子使用6-31G**基组. 出于计算耗时和精度考虑, 载体ZSM-5 分子筛计算模型截取直筒形双5T元环[20-22], 如图1(a)所示. 理论研究表明ZSM-5中T12位活性性质独特, 硅铝取代后得到的结构最优[17,23-25], 该载体中T9、 T12位上进行硅铝取代, 使其带有2个负电荷, Si、 Al上悬空键由H饱和, Si-H键长0.1460 nm, O-H键长0.1000 nm, 为保持分子筛构型稳定, 计算过程中, 所有Si-H和O-H键处于固定状态, 这模拟了沸石框架有限的刚性[20,26].(MnO)2+通过桥氧负载在ZSM-5上, 整个体系呈中性,模型见图1(b)[20]. 模拟过程中所有过渡态结构均通过IRC验证.

图1 优化后结构Fig.1 Optimized structure(a)ZSM-5; (b)Z2-[MnO]2+
反应气体分子的吸附能如式(3)所示:

式中Emolecule/cluster为带有吸附分子的团簇的能量,Ecluster为催化剂Z2-[MnO]2+的能量, Emolecule为吸附分子NH3、 NO等的能量.
2 结果与讨论
2.1 气体分子在Mn/ZSM-5催化剂上的吸附
进一步优化了气态NH3和NO分别在Z2-[MnO]2+模型上的吸附, 具体结果如图2所示, 分别记为Z2-[MnO-NH3]2+和Z2-[MnO-NO]2+, 计算了吸附后的吸附能(Eads, kJ/mol)、 键长(d, nm)和Mulliken(q, e)电荷, 具体结果如表1所示. 计算结果说明: NH3通过N端吸附于Z2-[MnO]2+上, Mn-N键长0.202 nm,吸附放热243.69 kJ/mol. 电子由NH3向催化剂表面转移的数量为0.246 e, N原子和H原子的相互作用由于失去电子而变弱, N-H键的键长发生微量的伸长, N-H键被活化. NO通过N端在Z2-[MnO]2+上吸附, Mn-N键长0.169 nm, 吸附放热210.09 kJ/mol. 电子由NO向催化剂表面转移数量为0.128 e, 失电子后, N原子和O原子的相互作用减弱, N-O键(0.115 nm)发生微量增长至0.117 nm, 被活化, 但吸附强度低于NH3. 因此, 在Z2-[MnO]2+催化剂表面,NH3更易吸附活化.
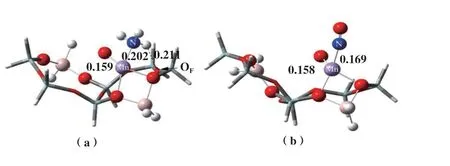
图2 优化后的分子吸附模型Fig.2 Optimized molecularadsorption model(a) Z2-[MnO-NH3]2+; (b) Z2-[MnO-NO]2+
2.2 NH3-SCR反应机理在Mn/ZSM-5催化剂上的研究
根据表1中气体分子的吸附结果可知, NH3在催化剂上的吸附强度略高于NO, 则可认为在Z2-[MnO]2+上起始步骤为NH3的吸附. 基于此, 我们分别遵循E-R、 L-H机理对Z2-[MnO]2+上的脱硝反应机理进行探究.

表1 NH3、 NO分别在Z2-[MnO]2+吸附后的键长(nm)、 Mulliken电荷(q, e)和吸附能(kJ/mol)Table1 Bond length (d, nm), Mulliken charge (q, e) and adsorption energy (Eads, kJ/mol) of NH3 and NO after Z2-[MnO]2+ adsorption
2.2.1 遵循E-R机理的路径1
按照E-R机理设计并完善反应路径1, 即: NH3-(ads)→ NH2(ads)→ NH2(ads)+NO(g)→ NH2NO-(ads) → NHNOH(ads) → N2+H2O, 涉及的基元反应如式(4)-(9)所示:

式(6)中NO气体分子直接与[NH2]反应, 图3为Z2-[MnO]2+上遵循E-R机理的路径所涉及到的所有物种的优化结构, 图4是此反应路径对应的能垒图, 表2是各基元反应的反应热和能垒.
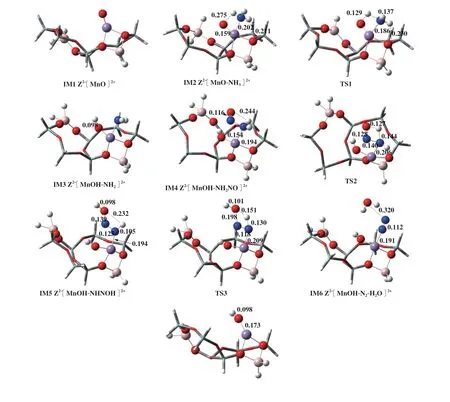
图3 遵循E-R机理的反应路径1Fig.3 Reaction path 1 following E-R mechanism

图4 遵循E-R机理的反应路径1的能垒图Fig.4 Energy barrier diagram of reaction path 1 following E-R mechanism
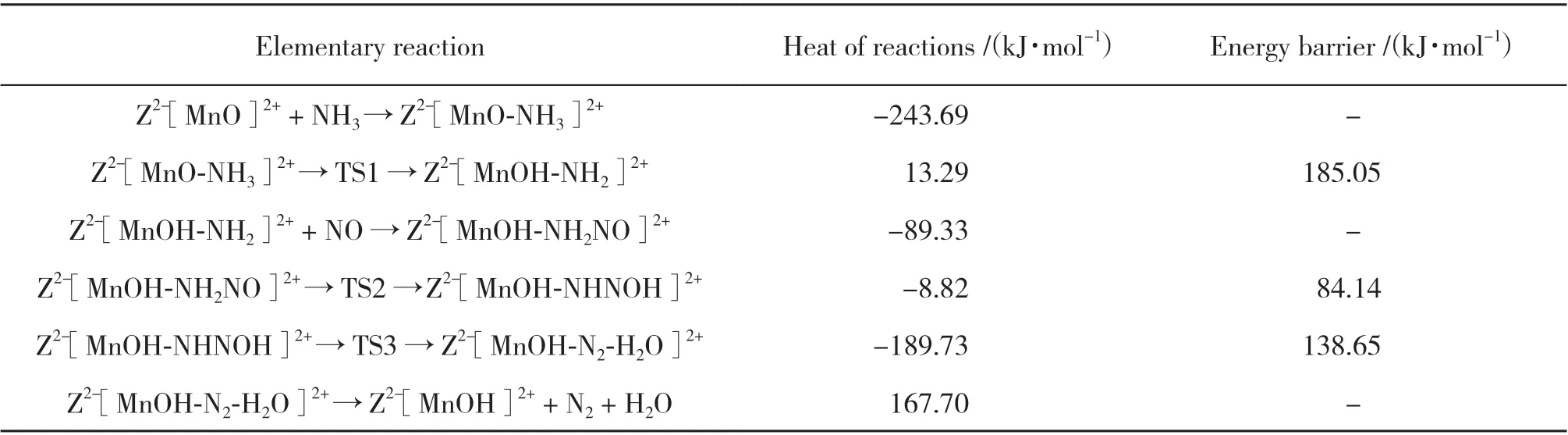
表2 遵循E-R机理的反应路径1的所有基元反应Table 2 All elementary steps of reaction path 1 following E-R mechanism
反应始于NH3分子的吸附, NH3以N端吸附于Mn上, 形成稳定构型Z2-[MnO-NH3]2+(IM2), NMn键长0.202 nm, 放热243.69 kJ/mol. [NH3]中一个H原子经过过渡态TS1转移至末端氧上形成Z2-[MnOH-NH2]2+(IM3), 其中N-H键距离由0.102拉长至0.137最后为0.351 nm. 而末端O与H形成OH键, 其距离由起始的0.275缩短至0.129 nm, 最终缩短为0.098 nm, 该过渡态反应能垒为185.05 kJ/mol. 随后与气相中NO分子反应, 生成N-N键, 键长0.154 nm, 形成重要中间体Z2-[MnOH-NH2NO]2+(IM4). 在氢键作用下, 中间体Z2-[MnOH-NHNOH]2+(IM5)和Z2-[MnOH-N2-H2O]2+(IM6)相继形成, 因氢键作用的存在, 其结构稳定, 通过过渡态TS2和TS3,N-H键断裂, O-H键生成, 两步基元反应需克服的反应能垒分别为84.14和138.65 kJ/mol, 形成的N2和H2O吸附在催化剂表面. 最后Z2-[MnOH-N2-H2O]2+体系吸收167.70 kJ/mol热量后, N2和H2O脱附, 形成Z2-[MnOH]2+(IM7).
2.2.2 遵循L-H机理的路径2
根据L-H反应机理设计并完善反应路径2, 即:NH3(ads) → NH3(ads)+NO(ads) →NH2NO(ads) →NHNOH(ads) → N2+H2O, 以 分 析Z2-[MnO]2+上 的SCR反应机理, 涉及的基元反应如式(10)-(15)所示:

式(11)中NH3和NO共同在Z2-[MnO]2+上进行吸附, 明显不同于反应路径1中气相NO直接参与反应, 并影响后续反应中的构型. 图5为Z2-[MnO]2+上遵循L-H机理的路径所涉及到的所有物种的优化结构. 反应路径2中所涉及到的系列稳定结构包括:反应物、 过渡态、 反应中间体和产物, 图6是此反应路径对应的能垒图, 表3是各基元反应的反应热和能垒.
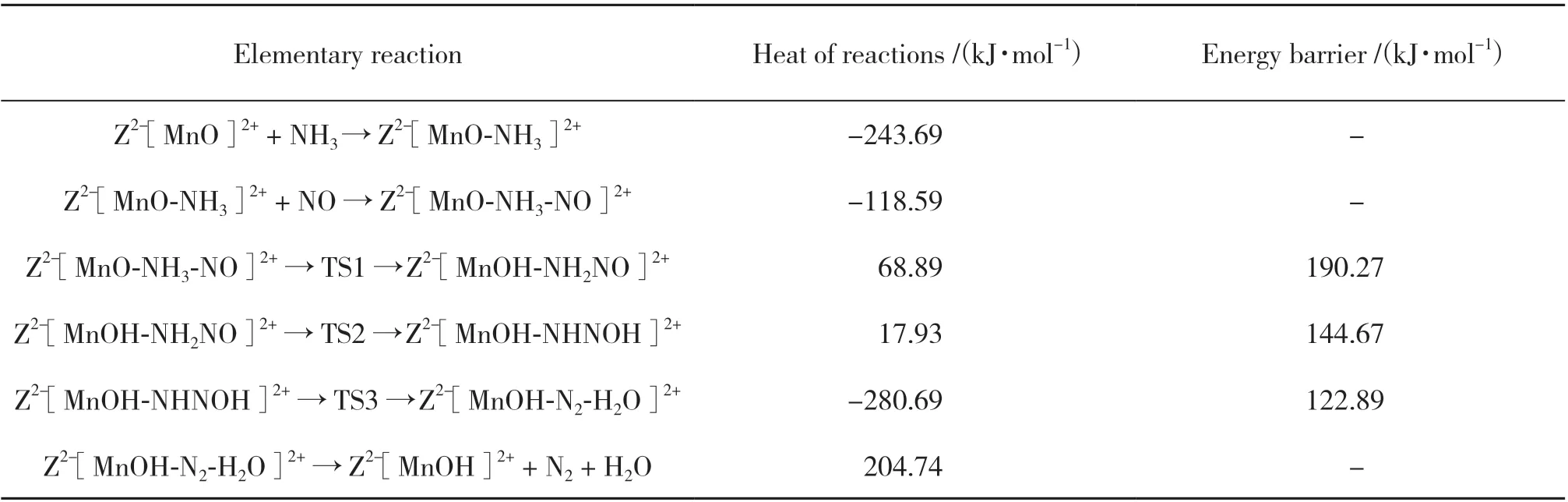
表3 遵循L-H机理的反应路径2的所有基元反应Table 3 All elementary steps of reaction path 2 following L-H mechanism

图5 遵循L-H机理的反应路径2Fig.5 Reaction path 2 following L-H mechanism
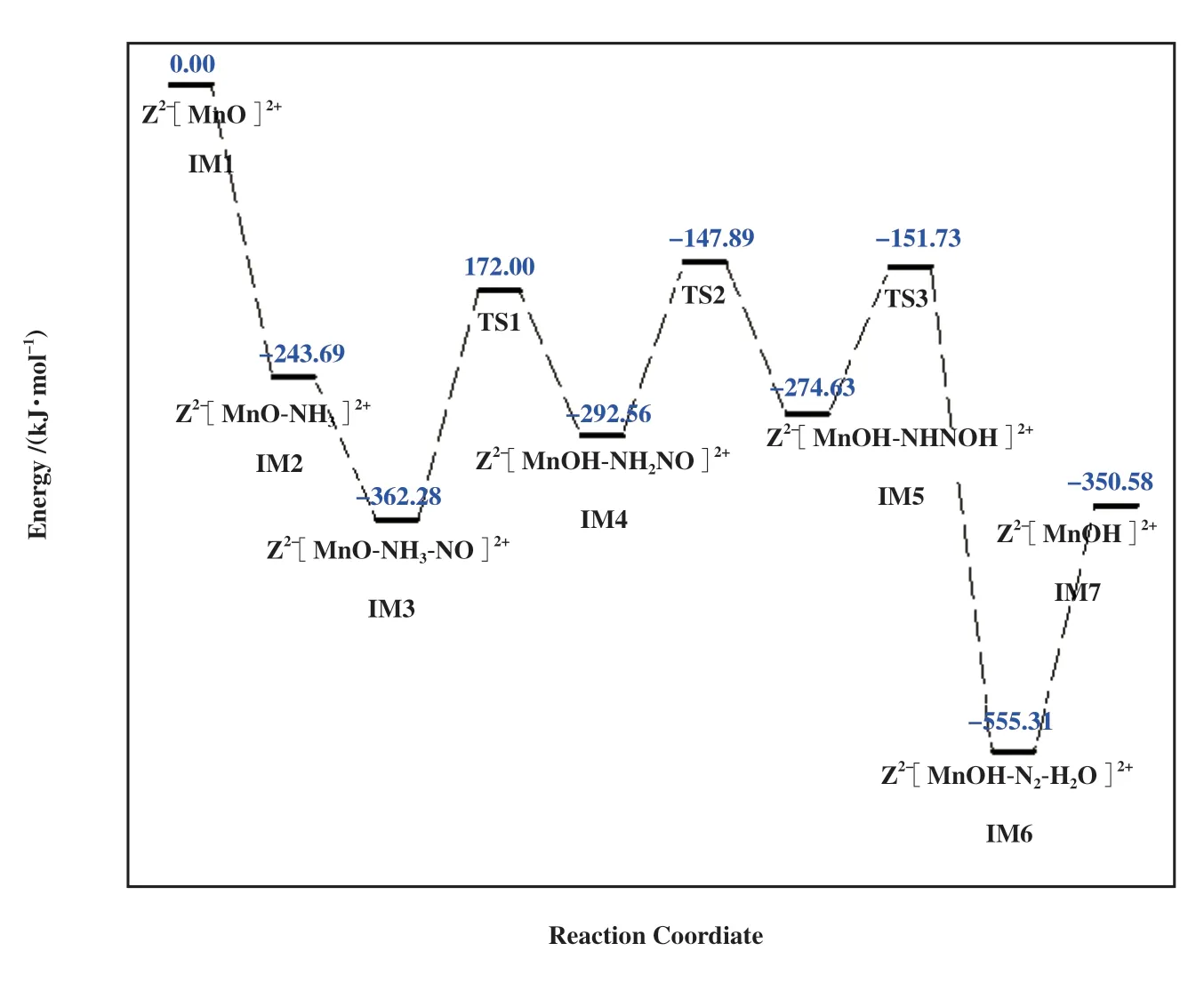
图6 遵循L-H机理的反应路径2的能垒图Fig.6 Energy barrier diagram of reaction path 2 following L-H mechanism
反应以NH3分子的吸附为起始步骤, NH3通过N端吸附于Mn上, 形成Z2-[MnO-NH3]2+(IM2), NMn键长0.202 nm, 放热243.69 kJ/mol. 然后NO通过N端在Mn位上吸附生成Z2-[MnO-NH3-NO]2+(IM3),其中Mn-N键长0.170 nm, 放热118.59 kJ/mol. 另外,NO可能通过O端吸附在Mn上, 但放热较少, 形成的结构不稳定, 不易与NH3反应, 因此NO选择以N端吸附. 通过过渡态TS1形成重要中间体Z2-[MnOHNH2NO]2+(IM5), 反应能垒为190.27 kJ/mol, [NH3]中N-H断裂, 键长由0.103伸长至0.153最后为0.301 nm. 而末端O与H的距离由0.281缩短至0.107最后为0.097 nm, 形成O-H键. 虽然该能垒较高, 但NH3和NO的吸附放出大量热量, 有利于该反应的进行. 通过过渡态TS2和TS3, 中间体Z2-[MnOH-NHNOH]2+(IM5)和Z2-[MnOH-N2-H2O]2+(IM6)相继形成. 反应能垒分别为144.67 和122.89 kJ/mol, NH2NO中的两个H相继和O接近, 继而发生N-H键的断裂, 生成O-H键, 反应产物N2和H2O都吸附于Mn上. 与反应路径1 相同, 过渡态TS2 和TS3 中形成类四元环结构, 稳定性强. 最后Z2-[MnOH-N2-H2O]2+体系吸收204.74 kJ/mol热量后, N2和H2O脱附, 形成Z2-[MnOH]2+(IM7).
3 结论
研究了NH3-SCR在Z2-[MnO]2+分子筛催化剂表面的反应机理. 通过密度泛函理论模拟计算了可能存在的两种反应路径, 分别遵循E-R机理和L-H机理, 得出以下主要结论:
1. 相较NO, NH3在Z2-[MnO]2+上的吸附结构最稳定, 因此在脱硝反应中易优先发生;
2. 路径1遵循E-R机理, NO气体分子直接参与反应, 与[NH2]形成中间体[NH2NO], 经过构型变化形成中间体[NHNOH], 最终分解为N2和H2O, 该反应过程中速控步为Z2-[MnO-NH3]2+→Z2-[MnOHNH2]2+, 反应能垒为181.20 kJ/mol;
3. 路径2遵循L-H机理, NH3与NO分子都先吸附在催化剂表面, 活化后的吸附态[NO]与[NH2]反应形成中间体[NH2NO], 后续机理与路径1相近,该反应过程中速控步为Z2-[MnO-NH3-NO]2+→Z2-[MnOH-NH2NO]2+, 反应能垒为186.97 kJ/mol;
4. 反应路径1中的速控步及其他基元反应需要克服的能垒相对较低, 则Z2-[MnO]2+上NH3-SCR反应更倾向于遵循E-R机理. 反应路径2中速控步的能垒较高, 需依靠NH3和NO的吸附热量来克服, 则低温下Z2-[MnO]2+催化剂上NH3-SCR反应易通过L-H机理进行. 同时遵循E-R机理和L-H机理的反应路径的速控步骤能垒相近, 在一定温度下可以同时遵循两种机理进行反应.
