不同组分过渡金属氧化物催化剂对介质阻挡放电固氮的影响机制
2022-11-15刘坤尹远耿文强夏昊天李华
刘坤,尹远,耿文强,夏昊天,李华
(1.重庆大学电气工程学院,重庆 400044;2.桂林电子科技大学生命与环境科学学院,3.广西自动检测技术与仪器重点实验室,桂林 541004)
低温等离子体固氮[1](Non-thermal Plasma Nitrogen Fixation,NTPNF)是近年来一种新型的绿色节能固氮技术,可在室温和大气压的条件下稳定持续地电离空气生成低温等离子体,从而得到氮氧化物(Nitrogen oxides,NOx).相较于传统的生物固氮[2,3],低温等离子体固氮具有过程可控、自由度高等优势.与传统工业固氮相比[4~6],则无需苛刻繁琐的高温高压反应条件,可减少能源的消耗和温室气体的排放[6],故采用低温等离子体固氮技术具有重要的理论研究价值和应用前景.
在低温等离子体的产生方式中,介质阻挡放电(Dielectric Barrier Discharge,DBD)因放电均匀度高、电子能量高、装置易于规模化、可与催化剂协同使用等特点而备受关注[7,8].国内外研究者围绕提高DBD低温等离子体固氮量[9~16]已开展了一些工作.如,Jogi等[9]研究表明DBD中可均匀地生成活性氧自由基(ROS),从而促进NOx的氧化反应并提高固氮量.Tang等[10]发现DBD中诸如ROS,N2等不同活性物质分别加速了NOx的产生或转化,使得DBD在最佳的能量密度下可获得最高的NOx产量;Pei等[11]以DBD作为等离子体产生方式,发现当功率密度提高时,NOx产率随之提升.研究发现,仅通过调控DBD参数不足以提高固氮产物NOx的转化率,同时固氮效率也有待提升,因此,研究人员采用了在DBD放电空间中填充过渡金属氧化物催化剂[12]的方式来进行优化,该类催化剂具有独特的价格优势以及良好的催化活性,能使等离子体电场和放电类型发生改变,进而改善固氮性能[12,13].Patil等[14]通过在DBD反应器中加入不同粒径(160~250 μm)的γ-Al2O3,测试了以其作为载体,所负载质量分数为5%的Co3O4或MoO3或V2O5等过渡金属氧化物催化剂对于DBD的影响,相比未填充催化剂的实验组,NOx的产率最多提升了100%.Chen等[15]发现在DBD反应器中填充WO3等n型半导体金属氧化物催化剂,其将与O2-和·OH等活性物质共同促进NO和NO2到N2O5的深度氧化;Cao等[16]研究了同轴DBD中填充催化剂的影响,发现Al2O3因较高的孔隙体积而具有较高的NO2生成率,而在Al2O3上负载MnOx则有更好的效果.究其原因,以Mn,Co,Mo,W和V等元素为代表的过渡金属氧化物催化剂,因其正负离子的缺位促成了特定的活性中心,伴随着价态的改变与能量的转移而具有一定的活性[17].这些催化材料沉积在具有较大比表面积的催化剂载体(如γ-Al2O3)上,为等离子体反应赋予了更多的活性氧,使得NOx产量得以提升.
现阶段,对于催化剂与DBD产生协同作用的机理研究还不够深入,通过填充多种活性组分的催化剂来提升DBD固氮效果的研究目前也较少,如何调控各活性组分的比例,进而优化等离子体固氮性能还值得进一步的探索.因此,本文采用可填充过渡金属氧化物催化剂的同轴结构DBD装置,通过高压交流电源驱动进行空气低温等离子体固氮反应,分别通过傅里叶变换红外光谱仪和紫外分光光度计对反应的气相产物和固定于液相的总氮进行了测定分析,并结合X射线衍射(XRD)、扫描电子显微镜(SEM)和X射线能谱(EDS)等表征手段,探究了引入催化剂对DBD固氮过程的影响机理,从而为低温等离子体协同催化固氮提供了一定的理论基础.
1 实验部分
1.1 试剂与仪器
活性氧化铝(γ-Al2O3,郑州金邦环保科技有限公司);六水合硝酸锰[Mn(NO3)2·6H2O]、六水合硝酸钴[Co(NO3)2·6H2O]和偏钨酸铵[(NH4)6H12W12O40],均为A.R.级,购于上海麦克林生化科技有限公司].
高压交流电源(自制);P6015A型高压探头和MDO 702型数字示波器(美国Tektronix公司);GA-83X型空气压缩机(上海硅莱实业有限公司);D07-7型质量流量计(北京七星华创电子股份有限公司);JC9023A型电热鼓风干燥箱(重庆松朗电子仪器有限公司);BF1200-A型高温箱式马弗炉(上海微行炉业有限公司);UV2200型紫外分光光度计(UV,北京瑞利分析仪器有限公司);Nicolet iS50型傅里叶变换红外光谱仪(FTIR,美国ThermoFisher公司);X′Pert Powder粉末X射线衍射仪(XRD,荷兰PANalytical公司);Quattro S型环境扫描电子显微镜(SEM,EDS,美国ThermoFisher公司).
1.2 负载型催化剂的制备
采用常规浸渍法制备催化剂:称取10 gγ-Al2O3作为催化剂载体,将其加入烧杯中,以去离子水冲洗表面以洗去多余杂质,将烧杯置入烘箱中于100℃干燥12 h以充分蒸发表面的水分,烘干后移入石英坩埚,置于马弗炉中于450℃烧结3 h,得到活化后的催化剂载体γ-Al2O3.按10%的质量分数称取催化剂原料,加入5 mL去离子水,配置成各自活性组分的前驱体溶液,搅拌振荡以充分溶解.将活化后的载体和溶液一并倒入烧杯,于室温下超声浸渍1 h.浸渍完毕后,先后经过100℃干燥12 h、500℃烧结3 h等步骤,制得单一型催化剂,分别记为Mn/γ-Al2O3,Co/γ-Al2O3和W/γ-Al2O3.
负载2种或3种活性组分的催化剂制备方法同上,在配制前驱体溶液时,按照10%的总质量分数和一定的活性组分配比来称取原料.如,Mn/Co/W等比例含量制备的三元复合型催化剂则是按照MnO2,Co3O4和WO3质量分数分别为3.33%称取原料制备而成.不同配比的二元复合型催化剂分别记为MnCo/γ-Al2O3,MnW/γ-Al2O3和CoW/γ-Al2O3;不同配比的三元复合型催化剂分别记为MnCoW/γ-Al2O3,Mn3CoW/γ-Al2O3,MnCo3W/γ-Al2O3,MnCoW3/γ-Al2O3.
1.3 DBD协同催化固氮实验过程
实验系统的示意图如Scheme 1所示,实验的气源为空气.以自制的带负反馈功能的高压交流电源输出交流高压施加于DBD装置,DBD装置采用水冷结构以避免放电时温度过高,与本课题组[18]之前实验所述结构一致,反应时的温度为350 K.通过在1 mm的DBD气隙中电离空气,从而产生不同的气相产物,气隙中可填充催化剂(每次填充量为2.0 g).气相产物的类别和浓度通过FTIR来识别和测定,波数范围500~2500 cm-1,而液相固氮结果则需将气相产物通入3个级联的玻璃洗气瓶中鼓泡吸收(洗气瓶中去离子水总体积为150 mL),实验时间为3 min,采用UV测定溶液的吸光度,再根据朗伯-比尔定律[19]将吸光度折算至总氮浓度(Total nitrogen concentration,TNC,以氮计,单位为mg/L).放电回路中串联一个电容来测量放电功率用以计算固氮能耗.实验前考察了1~8 L/min的空气气体流速对放电功率和固氮性能的影响,发现气体流量越大会导致空气的转化率降低,但是由于实验中流速对放电功率的影响可以忽略,此时固氮能耗会显著降低,因此实验中选取空气气体流速8 L/min为实验条件.

Scheme 1 Schematic diagram of the experimental systemMFC:mass flow controller.
2 结果与讨论
2.1 物相表征
为了确定催化剂的物相,对单一型催化剂进行了XRD分析,结果如图1所示.通过对照标准衍射卡片可以发现,3种催化剂均能在XRD谱图上呈现出各自金属氧化物的特征峰:MnO2(PDF#72-1984)在2θ=28.64°,37.31°,56.59°等处的衍射峰能够在图1(B)中很好地分辨出来,证明制备的Mn/γ-Al2O3的活性组分主要为MnO2;Co3O4(PDF#74-2120)在2θ=31.27°,36.85°,59.53°等处的衍射峰也能与图1(C)中Co/γ-Al2O3的XRD谱图相对应;W/γ-Al2O3在图1(D)中的XRD谱图,则与WO3(PDF#83-0947)在2θ=24.11°,34.07°等处的衍射峰重合度较高.此外,每组XRD曲线均有明显的单晶型γ-Al2O3衍射峰(在2θ=67.08°和37.05°等处),可对应图1(A)中所示的Al2O3的标准卡片(PDF#77-0396).这说明在负载了不同过渡金属组分的催化剂之后,载体γ-Al2O3的结构无明显差异,本实验中所用的Mn,Co和W 3种过渡金属元素分别能以MnO2,Co3O4,WO3的物相形式负载于催化剂载体γ-Al2O3之上.此外,在图1(B)和(C)中2θ=24.11°等处,还出现了几个明显的衍射峰.通过比对卡片PDF#74-1895,可知这几处衍射峰归因于催化剂上AlO(OH)的出现.在负载型催化剂的烧结过程中,由于Mn和Co的原料中含有结晶水,载体Al2O3会结合这些结晶水形成AlO(OH),因此在图1(B)和(C)中形成了AlO(OH)的衍射峰.而未负载的Al2O3以及负载W时原料中均不含有结晶水,因此,未在图1(A)和(D)中形成AlO(OH)的衍射峰.
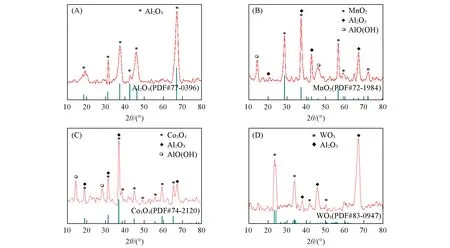
Fig.1 XRD patterns of catalysts γ-Al2O3(A),Mn/γ-Al2O3(B),Co/γ-Al2O3(C)and W/γ-Al2O3(D)
2.2 形貌表征
使用SEM观察了负载催化剂的表面形貌(图2),其中图2(A)~(D)分别为载体γ-Al2O3以及负载型催化剂Mn/γ-Al2O3,Co/γ-Al2O3和W/γ-Al2O3的结果.从图2(A)中可以看到,载体的表面有较多的小颗粒,具有丰富的孔隙结构,这使得载体γ-Al2O3具有较大的比表面积,有助于提升催化活性.而在图2(B)~(D)中,分别于载体上负载MnO2,Co3O4和WO3等活性组分之后,氧化物在载体表面的堆积使得孔隙结构更加丰富,其中Mn/γ-Al2O3的颗粒较大,Co/γ-Al2O3的颗粒较小,而W/γ-Al2O3的表面则多为凸起的团簇状结构,这些都在一定程度上增加了活性粒子在催化剂表面的吸附位点,当被填充于DBD放电气隙中时,将有更高的概率与空气等离子体产生协同作用,从而促进催化活性.

Fig.2 SEM images of catalysts γ-Al2O3(A),Mn/γ-Al2O3(B),Co/γ-Al2O3(C)and W/γ-Al2O3(D)
2.3 元素分布表征
为了确定复合型催化剂的负载情况,对不同配比的三元复合型催化剂进行了EDS分析,运用仪器的面扫描功能(Mapping)得到催化剂表面的元素分布.图3(A)~(C)所示分别为Mo/Co/W质量比为3∶1∶1,1∶3∶1和1∶1∶3的三元复合型催化剂的EDS谱图,表1所示为不同催化剂对应的各元素所占比例.从图表中可以发现,除了作为载体的Al2O3占据了较多的Al和O元素,催化剂上的Mn,Co,W元素均能够按照投料配比较好地分布于载体表面.如图3(A)中,Mn在5.89 eV处的特征峰强度较Co(6.93 eV)和W(8.41 eV)处更为突出,且测得的Mn/Co/W元素的质量比接近3∶1∶1,同理,图3(B)和(C)中的结果也分别接近1∶3∶1和1∶1∶3,这说明复合型催化剂的制备较符合设计预期.
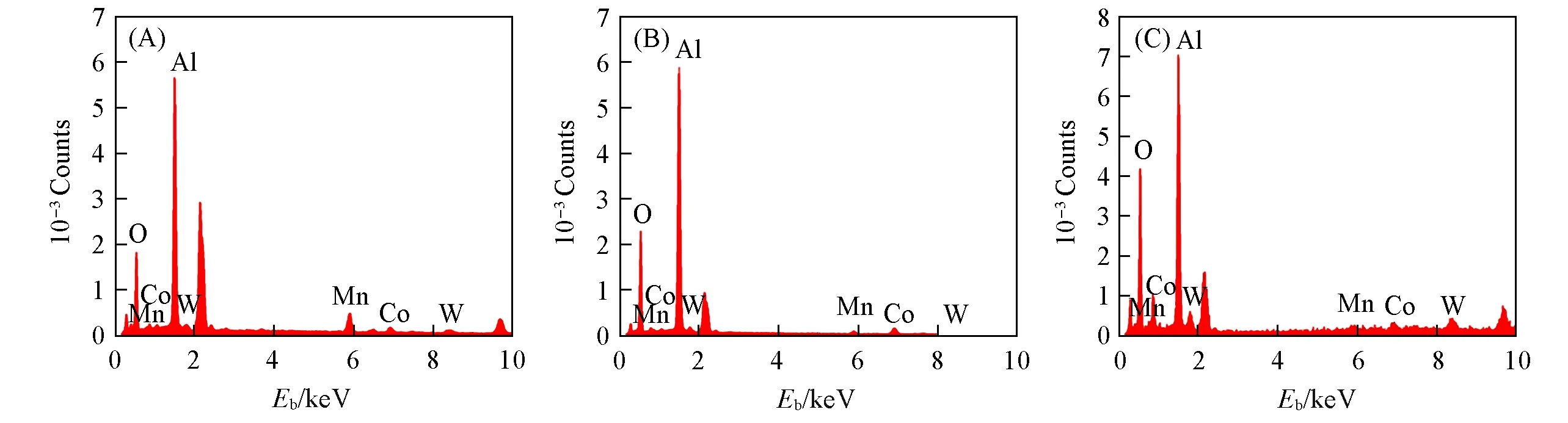
Fig.3 EDS spectra of trinary catalysts Mn3CoW/γ-Al2O3(A),MnCo3W/γ-Al2O3(B),MnCoW3/γ-Al2O3(C)
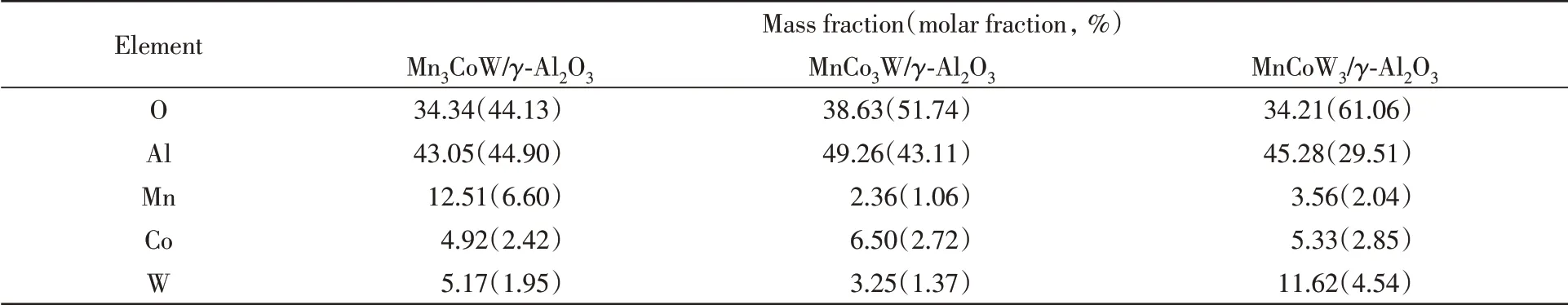
Table 1 Elements ratios in different three-way catalysts
2.4 DBD协同催化固氮性能
以总氮浓度(TNC)随电压的变化曲线示出了填充不同催化剂的DBD协同催化固氮性能,TNC随着电压的升高均呈现出先增大后减小的趋势.
填充单一型催化剂的协同催化固氮性能如图4(A)所示.可以看出,仅填充载体γ-Al2O3时对提升TNC效果甚微,其结果与未填充催化剂组相近.而在负载了活性组分后,各组的TNC均有所提升,并且曲线的极值点向电压更高的方向略有偏移.其中,负载Mn/γ-Al2O3的固氮效果最佳,在20 kV处取得最高的TNC(102.25 mg/L),比未填充催化剂组在18 kV处取得的最高值(69.42 mg/L)提高了47.30%.W/γ-Al2O3组协同固氮效果次之,在20 kV处取得最高的TNC(91.48 mg/L);Co/γ-Al2O3组协同效果最差,在22 kV处取得最高的TNC(81.80 mg/L),但是相比于未填充催化剂组的最大值也提高了17.83%.相应地,Mn/γ-Al2O3,W/γ-Al2O3,Co/γ-Al2O33种一元催化剂的最高单位催化剂产率分别为153.34,137.22和122.7 mg·g-1·h-1.DBD装置中填充催化剂会对放电产生影响,进而影响放电功率和固氮性能.实验发现,无填充、γ-Al2O3上无负载活性组分及负载Mn,W或Co时,在固定电压20 kV时所对应的放电功率分别为163.51,166.02 W和173.27,172.56,172.47 W.结合图4(A)的结果可以发现,在相同实验条件下,γ-Al2O3上无负载活性组分时,放电功率与无填充时差别不大,因此仅填充载体γ-Al2O3时对提升TNC效果甚微;而γ-Al2O3上负载活性组分后放电功率显著提高,导致各组的TNC均有所提升.但是负载活性组分后各组的提升幅度不一致,这主要受到活性组分性质的影响.
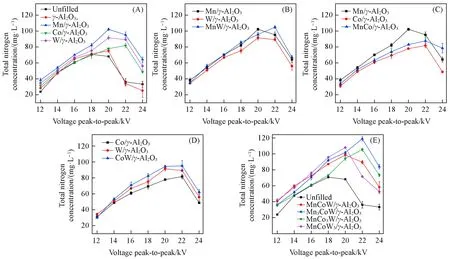
Fig.4 Variations of TNC with voltages when filled with single catalysts(A),binary catalysts(B—D)and ternary catalysts(E)
填充二元复合型催化剂的协同催化固氮性能如图4(B)~(D)所示,与单一活性组分的固氮性能进行对比,发现二元复合型催化剂组与单一型催化剂组的TNC结果相近,且极大值均处于电压为22 kV处.其中MnW/γ-Al2O3组取得最高为104.90 mg/L的TNC值,高于Mn/γ-Al2O3组和W/γ-Al2O3组TNC的最高值;MnCo/γ-Al2O3组取得最高87.67 mg/L的TNC值,低于Mn/γ-Al2O3组的TNC最高值,高于Co/γ-Al2O3组的TNC最高值;CoW/γ-Al2O3组则取得最高95.30 mg/L的TNC值,高于Co/γ-Al2O3组和W/γ-Al2O3组的TNC最大值.相应地,MnW/γ-Al2O3,MnCo/γ-Al2O3,CoW/γ-Al2O33种催化剂的最高单位催化剂催化产率分别为157.35,131.51和142.95 mg·g-1·h-1.
三元复合型催化剂的协同催化固氮性能如图4(E)所示,除了MnCo3W/γ-Al2O3实验组在电压为14~18 kV时固氮优化效果较弱,TNC接近未填充催化剂组以外,其余各条件下三元复合型催化剂的TNC相比未填充催化剂组均有一定提高.以Mn3CoW/γ-Al2O3组在电压为22 kV处取得的TNC最大值119.13 mg/L计算,其在单一型催化剂的基础上再度提升了18.08%,相比于未填充催化剂组的最高值提升了71.61%.相应地,Mn3CoW/γ-Al2O3组的最高单位催化剂催化产率为178.70 mg·g-1·h-1.利用李萨如图法计算放电功率时,发现未填充催化剂组在取得最高TNC值(69.42 mg/L)的18 kV时的放电功率为137.48 W,填充Mn3CoW/γ-Al2O3实验组在取得最高TNC值(119.13 mg/L)的22 kV时的放电功率为184.71 W.实验组中放电功率增加是由于放电强度增强,放电电流增大导致的,此时未填充催化剂组和实验组对应的能耗分别为33.27和26.05 MJ/mol.Mn3CoW/γ-Al2O3实验组生成NOx的能耗比未填充催化剂组降低了21.70%.实验前后对比了Mn3CoW/γ-Al2O3催化剂的形貌特征变化,结果如图5所示.对比图5(A)和(B)可以看到,放电处理后催化剂表面颗粒尺寸变小,孔道结构变丰富.这是因为放电等离子体对催化剂具有改性效应,放电体系中的高能电子以及重离子等能轰击碰撞材料表面、促进孔结构重整.在等离子体催化剂协同固氮过程中,催化剂性质的实时变化可能会影响固氮效率,这需要后续更深入的研究来探讨催化剂实时变化的影响.

Fig.5 SEM images of Mn3CoW/γ-Al2O3 catalysts before(A)and after discharge(B)
等离子体气相产生的NOx需要进一步转化生成液相NOx才能被实际应用,而气相NOx不能完全转化为液相NOx.如,Dinh等[20]研究发现放电产生的气相NOx向液相NOx的转化率大概在20%左右.表2[21~23]比较了不同放电装置生成液相NOx的产率和能耗,可以看到,自制的交流电源驱动DBD结构放电能在取得较好的液相NOx产率的同时具有较低的能耗.
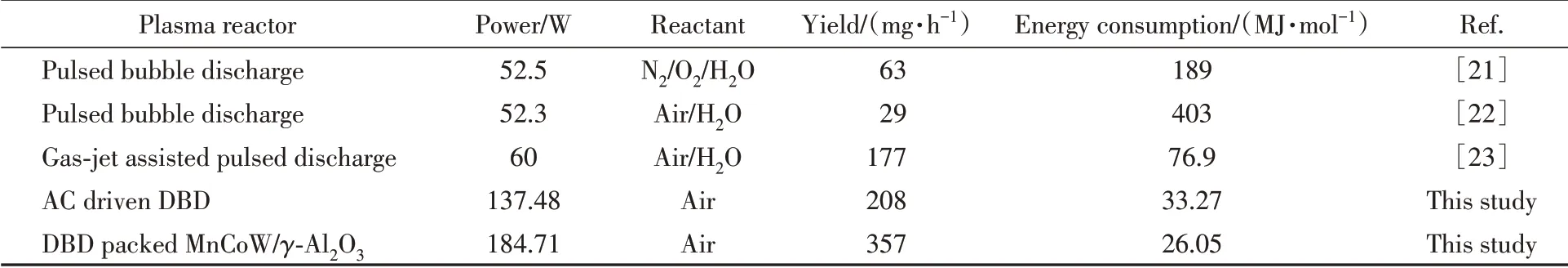
Table 2 Comparison of liquid NOx production rate and energy consumption with literatures
2.5 DBD气相产物的FTIR表征
NOx的生成是空气DBD氮氧反应中各种活性粒子共同作用的结果[24,25],然而并非所有的NOx对于固氮都有着积极的影响,NOx的溶解度会随着氮的价态升高而提高.N2O难溶于水且性质稳定;NO在水中的溶解度很低却易被氧化成NO2;NO2易溶于水生成HNO2/HNO3;NO3虽为不稳定的NOx组分中间体,却能与NO2结合生成溶解度最高的N2O5;N2O5则作为硝酸的酸酐可以直接被水吸收为HNO3并于液相中固定.为了能深入分析催化剂对产物的影响,采用FTIR对无催化剂和Mn3CoW/γ-Al2O3(产量最高)两种情况下的产物进行了测定.
图6为DBD气相产物随电压变化的FTIR谱图,图6(A)和(B)分别对应未填充催化剂和填充催化剂Mn3CoW/γ-Al2O3时的结果.从图6(A)中可以明显地观察到,随着电压由12 kV逐步上升至24 kV,O3的吸光度不断减弱,NO2吸光度不断增强,N2O在1300和2238 cm-1两处特征峰强度相较于NO2而言变化较小,并且在所有电压下均存在N2O的吸收峰,此外1720 cm-1处N2O5的特征峰强度随电压的升高先增加后降低,而NO的特征峰仅在未填充催化剂组当电压升至22和24 kV时才出现.图6(B)中几种气相产物特征峰强度的变化规律类似,但区别最明显之处在于NO2和N2O5对应的特征峰强度明显增高,且NO的特征峰消失.
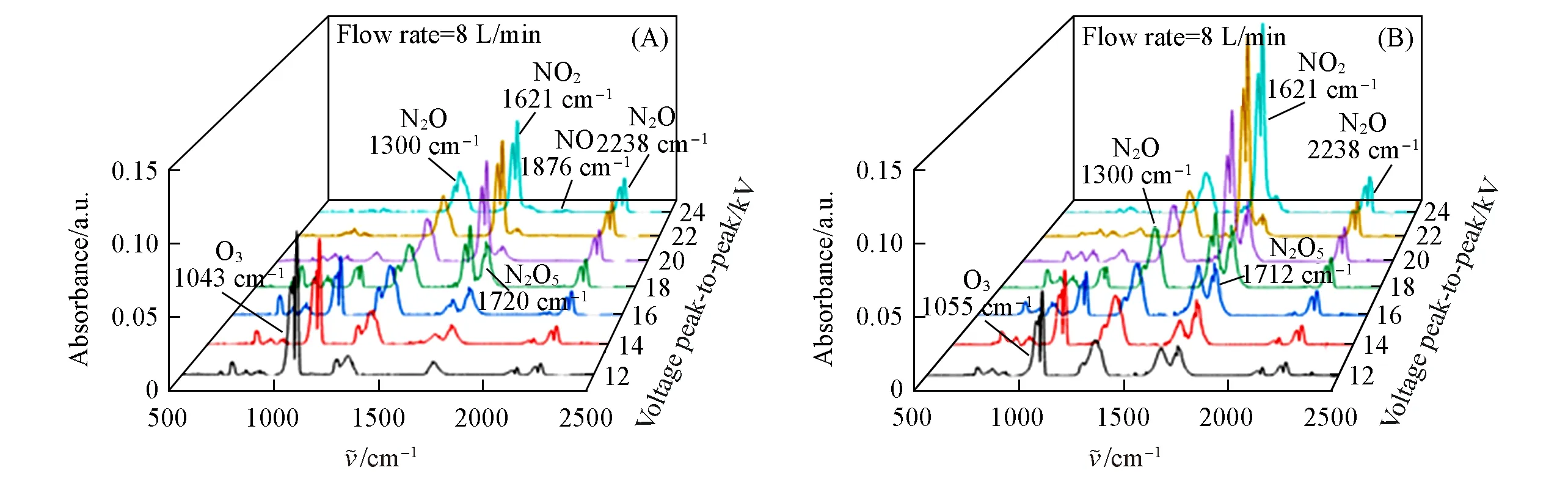
Fig.6 FTIR spectra with catalyst unfilled(A)or filled with ternary catalysts Mn3CoW/γ-Al2O3(B)
为了清晰反映出填充催化剂前后气相产物中不同种类NOx以及O3的浓度变化,将图6(A)和(B)中几种NOx特征峰处的吸光度以折线图的形式呈现于图7中,以电压峰-峰值为横轴,各气相产物的吸光度为纵轴.其中,O3取波数为1043 cm-1处特征吸收峰的吸光度,NO2,N2O5,NO和N2O则分别取波数为1621,1720,1876和2238 cm-1处特征峰的吸光度以作对比.从图7(A)和(B)中可以更加直观地观察出,虽然填充催化剂后各气相产物随电压的变化趋势基本同未填充催化剂时一致,但吸光度发生了一定的变化,如NO2和N2O5在填充催化剂后吸光度明显提高,这将直接起到提高液相总氮浓度的作用;NO在未填充催化剂组电压分别为22和24 kV时有非零的吸光度,而在填充催化剂后各电压下均接近于0,难溶性NO的减少也对促进固氮效率有益.

Fig.7 Absorbance of O3 and NOx varies with voltage of catalyst unfilled(A)and filled with catalyst(B)
2.6 DBD协同催化固氮机理
为了解释上述现象以及TNC出现高低的原因,将结合DBD气相化学反应来进行分析.Scheme 2展示了以空气为工作气体的DBD放电体系中主要的气相链式反应,由3个部分组成.表3给出了相关的气相反应式.Scheme 2的第Ⅰ部分表征了DBD放电体系中,高能电子与气体分子N2,O2发生碰撞、吸附、离解等反应,生成O,N,等活性粒子的过程[26,27],表3 Entries 1~10为部分相关的化学方程.式中的M为参与DBD反应的中性气体分子,如O2,N2,NO,NO2等.正是这些活性粒子在微观上氧化/还原过程的竞争,导致了宏观上整体气相反应过程的可逆性.
Scheme 2的第Ⅱ部分表示在这些活性粒子的氧化还原作用下,产生了NO,N2O,NO2,N2O5等NOx的过程[28,29],其中涉及到的部分反应如表3 Entries 11~26所示.由于氧气的离解能(5.12 eV)小于氮气(9.76 eV)[30],O2较N2更易电离.当电压较低时,DBD反应体系中以具有氧化性的O3,O-2,O等ROS为主,O3特征峰强度最高.ROS使得部分N2电离出的N很快便通过表3 Entries 11~18被氧化至具有最高氮元素价态的N2O5.此外,通过表3 Entry 5中N2受电子碰撞激发所产生出的N2(A3∑+u),是一种极具还原性的短寿命活性粒子,其生成早于N,一经生成就与DBD反应体系中的O2通过表3 Entry 19生成溶解度极低的N2O[31],N2O是一种很稳定的NOx,其结构保留了N2中的N≡N,甚至能与强氧化性的O3共存,它在NOx中的比重过大将严重影响固氮效果.

Scheme 2 Gas-phase chain reactions of DBD process

Table 3 Gas phase correlation reations
随着电压的提高,DBD的输入能量逐渐增大,N2加快离解出具有还原性的N,N(2D),N2(A3∑+u)等活性粒子[18],迅速与ROS通过表3 Entries 11~14中的反应生成NO2,这一过程表现为图6中NO2吸光度的增加和O3吸光度的减少.同时,NO2的生成提升了N2O5和N2O的吸光度,如表3 Entries 15~18和Entry 21所示,这进一步消耗了DBD放电空间内的ROS,使得NOx逐渐替代O3成为气相产物的主导.
当电压进一步提升,输入的能量继续增加,气相产物中除了NO2还检测到了1876 cm-1处NO的红外吸收峰.NO的生成意味着此时DBD反应体系中不存在具有强氧化性的O3,如图7(A)所示,O3吸光度的下降进一步促进了NO2和N2O的生成反应.除了O3,N2O5的吸光度在电压从18 kV升至24 kV的过程中也迅速减弱,这是由于O3的消耗使NO3的生成受到限制,同步减弱了N2O5的生成(表3 Entry 18),且N2O5和NO3本身不如NO2稳定,易通过表3 Entries 22和23重新转化为NO2.作为溶解度最高的NOx,N2O5吸光度的降低是导致图4中出现电压继续提升后TNC下降现象的直接原因.
对于催化剂固氮性能的分析可拆解成载体和活性组分两个方面.首先,对于载体γ-Al2O3,其本身在等离子体过程中并不够活跃,它主要是使得具有活性的过渡金属氧化物活性组分均匀地在其表面分布,提供较高的比表面积以提高催化反应的活性[14].图4(A)中,仅填充了载体γ-Al2O3的实验组与未填充催化剂组的TNC大小以及TNC随电压变化的趋势相近,这说明仍然需要活性组分与DBD产生耦合协同作用来提高固氮效果.
其次,对于活性组分,其组成为过渡金属氧化物,从图1的XRD结果可见,金属氧化物并未与Al2O3烧结反应形成新的化学键或物质,仍以氧化物形式负载于γ-Al2O3上.负载活性成分的催化剂填充于DBD放电气隙中能够提供出更多的氧空位[32],加速NOx的进一步氧化.表3 Entries 24~32反映了这一过程,式中:M代指过渡金属元素;*表示催化剂的活性位点;(ads)表示吸附态.表3 Entries 24~32表明,催化剂上的活性位点能够吸附DBD过程中长寿命或短寿命的活性粒子,通过O3在催化剂表面分解出O形成吸附态M-O(ads),促进增强催化剂表面NOx的逐步氧化(表3 Entries 29~31),如此使得产物中可溶性氮氧化物的成分增多,提升了固氮的效果.
催化剂表面吸附-氧化的过程中同时伴随着活性金属元素的价态变换和能量转移,即Scheme 2中DBD气相链式反应体系的第Ⅲ部分.本文中3种金属元素各有特点,如Mn能够提供晶格氧[33]并在其表面产生大量氧空位,促进NO到NO2再到N2O5的反应,而Mn元素在催化氧化NOx的过程中经历+4/+3/+2价态的变换,同时等离子体产生的ROS很容易将其再氧化至+4价,保证了其催化稳定性,本实验中含Mn催化剂的实验组均有着较高的固氮量提升(图4);W作为n型催化剂常见于光催化领域,在DBD协同固氮实验中,以氧化钨稳态WO3和过渡态WO3-x[34]的形式置于DBD放电空间,同样由于高氧化还原电位而为固氮反应提供了活性位点及赋能;Co以+2和+3价变换,与Mn和W相比,Co基实验组固氮效果稍有不足,这是由于Co3O4是一种典型p型半导体金属氧化物,而MnO2和WO3为n型[35],n型半导体的多为电子而p型半导体为空穴,DBD过程中会激发出高能电子,从而有助于提升n型催化剂的催化活性,促进表面NOx的吸附氧化作用,本文中3种元素催化剂的DBD催化固氮性能均优于未填充催化剂组和仅填充载体γ-Al2O3组,除了过渡金属的吸附氧化作用之外,填充催化剂将使得反应空间紧缩,每个气体分子获能活化的概率得到了提升,等离子体协同固氮反应更加充分,这是固氮效果提升的另一个原因.
最后,在单一型催化剂的基础上,负载多类过渡金属氧化物活性组分构成多元复合型催化剂,可加强金属元素间的相互作用,减少团聚效应从而提升催化剂的分散性[36].此外,多种活性组分催化剂的填充,将使得不同过渡金属离子之间发生电子转移,使价态变换更为频繁,进而提升DBD协同催化过程中能量的传递,促进催化剂的活性和稳定性.本文三元复合型催化剂Mn3CoW/γ-Al2O3测得总氮浓度最大,可由上述理论解释如下:在Mn基催化剂中掺入Co和W元素,将引起DBD过程中催化剂表面以及等价态的循环变化,且Mn有7个价电子,价态变换较另外两种催化剂多,更具氧化活性[31],在DBD协同催化固氮反应中能提供更多的活性氧空位,在保持催化剂价态宏观不变的同时,3种组分相互耦合,加速了DBD放电空间中能量的转换,进而提升了单位时间内对气相产物NOx的氧化作用以及固氮效率.
3 结论
制备了不同过渡金属氧化物作为活性组分的负载型催化剂,通过将催化剂置于DBD放电空间内进行低温等离子体协同催化反应来提升固氮性能.采用紫外分光光度法测量了液相中的总氮浓度TNC,实验结果表明,填充了催化剂的实验组对比未填充催化剂组的TNC明显提升.TNC三元复合型催化剂Mn3CoW/γ-Al2O3具有最高的TNC值,可提升至119.13 mg/L,较未填充催化剂组提高了71.61%,此时能耗降低了21.70%.通过傅里叶变换红外光谱仪对有无填充催化剂的DBD气相产物进行了测定,结合DBD模式转换机理分析了电压提升导致NOx产量先升后降的原因在于NOx的氧化程度不够,并通过FTIR结果证明了填充催化剂能促进空间内NO2和N2O5的生成.通过空气DBD气相中链式反应机理和过渡金属氧化物催化机理揭示了固氮量得以提升,是因为催化剂在等离子体过程中提供了大量氧空位,这给予了NOx更多氧化的渠道.负载多种过渡金属元素的复合型催化剂在DBD协同催化固氮过程中通过价态的变化加速了能量的传递,使NOx氧化反应加快,进而促进了固氮效果.通过本研究,证实了多元过渡金属氧化物复合型催化剂在等离子体固氮领域对提高产量和降低能耗方面的优势,而且不同过渡金属氧化物之前的耦合效应具有差异,这为后续高效催化剂的设计提供了思路.此外,实验中发现放电等离子体对催化剂具有改性效应,探讨等离子体催化剂协同固氮过程中催化剂性质的实时变化如何影响固氮效率对优化催化剂和延长催化剂使用寿命具有重要意义.