基于上转换发光共振能量转移的CRISPR/Cas12a生物传感系统用于HPV16 DNA双信号检测
2022-11-15章丽玲刘浏郑明秋方文凯刘达唐宏武
章丽玲,刘浏,郑明秋,方文凯,刘达,唐宏武
(武汉大学化学与分子科学学院,武汉 430072)
光学传感和分析在医学领域具有重要作用,在众多体外生物检测方法中,发光生物检测法因其方便的光信号转导、高灵敏度和快速响应而成为目前主要的分析工具[1~4].传统的发光探针,如有机染料[5~7]、量子点[8]和金银纳米簇等[9~11],存在背景噪声高、紫外及可见光引起的样品光损伤、光漂白阈值低和潜在的毒性等缺点.基于镧系离子独特的化学和光学性质,稀土上转换纳米材料(UCNPs)具有稳定性高[12,13]、细胞毒性低和对生物样本几乎没有光损伤等优点[14~17].此外,UCNPs的单波长激发和多波长发射有利于多通路生物分子的测定,可以避免其它荧光的干扰,这些特性使掺杂Ln3+的UCNPs有望成为新一代发光生物探针.
在过去的几十年中,已开发了多种基于Ln3+掺杂的UCNPs的新型高效生物分子测定技术,这些生物传感系统的检测机制主要通过发光共振能量转移(LRET)过程实现[18].在典型的上转换-发光共振能量转移过程中,Ln3+掺杂的UCNPs受到近红外光激发时,会以非辐射的方式将能量转移给邻近的光谱匹配受体(通常在10 nm或更小范围内),导致UCNPs的发光猝灭[19].这些受体通常是可与UCNPs在峰值波长处发射匹配的吸收系数大的分子或纳米材料,如荧光蛋白、有机染料、量子点、金纳米颗粒或纳米棒、二氧化锰纳米片、石墨烯和石墨烯氧化物等[20~25].自Wang等[26]在2005年首次报道上转换-发光共振能量转移过程以来,已经开发了许多基于UC-LRET过程检测生物分子的方法.
成簇规律间隔的短回文重复序列(CRISPR)及其相关蛋白(CRISPR/Cas)系统是一种细菌用于抵抗外来遗传物质入侵的适应性免疫系统,主要由Cas和CRISPR RNA(CrRNA)组成,该系统中Cas蛋白可以对入侵的外源DNA进行特异性切割,从而达到免疫效果[27].Cas12a作为CRISPR/Cas系统的重要成员,通过识别PAM短序列的5′T,可以对dsDNA进行特异性切割(顺式切割);同时,Cas12a对ssDNA的切割(反式切割)活性也得到激发[28],利用Cas12a的这种反式切割活性可实现对其它核酸分子的传感检测.本文首先在常规核UCNPs(C-UCNPs)基础上合成了核/壳/壳内壳层发光结构UCNPs(CSS-UCNPs),可提高LERT效率(Scheme 1).随后,利用CRISPR/Cas12a系统对ssDNA的反式切割活性,基于AuNPs比色和UCNPs发光检测实现了对人乳头瘤病毒DNA(HPV16 DNA)的双信号灵敏检测.当目标物HPV16 DNA不存在时,CRISPR/Cas12a系统对Linker-ssDNA不发生切割,完整的Linker ssDNA可连接DNA-1/2-AuNPs,使二者发生团聚,溶液变为蓝紫色;当目标物HPV16 DNA存在时,可激活CRISPR/Cas12a系统的反式切割活性,切割Linker ssDNA,此时DNA-1/2-AuNPs由于无法发生连接,保持分散状态,溶液呈红色,通过比色法可初步完成定性检测;团聚程度不同的DNA-AuNPs在527 nm吸收强度不同,使其对UCNPs在542 nm发射峰的猝灭效果也不同,UCNPs的发光信号发生改变,通过聚乙烯亚胺(PEI)CSS-UNCPs发光信号的变化可以进一步实现对目标DNA的定量检测,比色法与荧光法双信号检测的结合有效提高了对目标分析物检测结果的准确性.
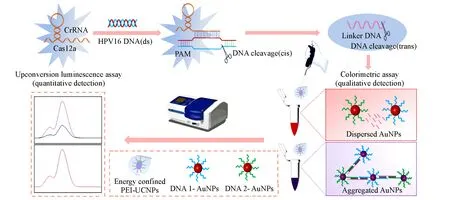
Scheme 1 Schematic illustration of analysis procedure for dual signal detection of HPV16 DNA by CRISPR/Cas12a biosensor based on upconversion luminescent resonance energy transfer
1 实验部分
1.1 试剂与仪器
YCl3·6H2O(纯度99.99%)、YbCl3·6H2O(纯度99.99%)、ErCl3·6H2O(纯度99.99%)、十八烯(C18H36,纯度95%)、油酸(C18H34O2,纯度90%)(OA)、氟化铵(NH4F)、三水合氯化金(HAuCl4·3H2O)、三(2-羧乙基)膦(TCEP)、二水合柠檬酸三钠(Na₃C₆H₅O₇·2H₂O)和聚乙烯亚胺(PEI,Mw=1800)均购自阿拉丁试剂公司;LbCas12a购自广东博徕斯生物科技公司;其它分析纯试剂均购于国药集团化学试剂有限公司;实验用超纯水电阻率为18.25 MΩ·cm.所用DNA序列购自生工生物工程(上海)股份有限公司,具体序列信息见本文支持信息表S1.
FTIR5700型傅里叶红外光谱分析仪(FTIR,美国Thermo公司);XPert Pro型X射线衍射仪(XRD,荷兰帕纳科公司);UV-2550型紫外-可见分光光度计(UV-Vis,日本Shimadzu公司);F-4700型荧光分光光度计(日本Hitachi公司);JEM-2100 Plus型200 kV高分辨透射电子显微镜和JEM-2100 F型200 kV高分辨透射电子显微镜(HRTEM,日本JOEL公司);Zetasizer Nano ZEN3600型动态光散射仪(DLS,日本Malvern公司);HC-2518R型高速冷冻离心机(安徽中科中佳公司).
1.2 实验过程
1.2.1 上转换纳米材料的合成在文献[29]报道的高温共沉淀法的基础上进行部分改进,通过介导壳层外延成长法合成核/壳/壳内壳层发光的上转换纳米材料(CSS-UCNPs).
向100 mL三颈烧瓶中加入1 mmol YCl3·6H2O、15 mL十八烯和6 mL油酸,搅拌混合后,将体系抽真空40 min;随后,在氩气保护下升温至160℃,加热40 min,白色固体完全溶解,得到淡黄色透明均相溶液;冷却至室温,缓慢匀速加入含有4 mmol NH4F和2.5 mmol NaOH的6 mL甲醇溶液,剧烈搅拌后形成白色浑浊液.随后,加热升温至110℃,反应30 min以去除体系中的甲醇,抽真空30 min以去除残存的甲醇和水分子,在氩气保护下快速升温至310℃,反应1 h后,向上述溶液加入20 mL乙醇,以12000 r/min转速离心5 min,所得白色沉淀经体积分数50%的乙醇洗涤3次后,再使用正己烷/乙醇混合溶液(体积比1∶3)洗涤3次,得到核UCNPs(命名为C1-UCNPs)并分散在4 mL三氯甲烷溶液中,置于4℃冰箱中用于后续合成.
向100 mL三颈烧瓶中加入n(Y)/n(Yb)/n(Er)=0.80/0.18/0.02的0.75 mmol稀土盐混合物、11.5 mL十八烯和4.5 mL油酸,将混合体系抽真空40 min;随后,在氩气保护下升温至到160℃,加热40 min至白色固体完全溶解,得到淡黄色透明均相溶液;冷却至室温,注射加入C1-UCNPs核(core),加热至110℃,反应30 min以去除体系中的氯仿,降至室温后匀速缓慢加入含有3 mmol NH4F和1.8 mmol NaOH的5 mL甲醇溶液,剧烈搅拌,形成白色浑浊液.后续步骤与之前相同,最后得到核/壳UCNPs(命名为CS-UCNPs)并分散在4 mL三氯甲烷中,置于4℃冰箱中用于后续合成.
向50 mL三颈烧瓶中加入0.25 mmol YCl3·6H2O、7.5 mL十八烯和3 mL油酸,将混合体系于室温抽真空40 min;随后,在氩气保护下升温至160℃,加热30 min至白色固体完全溶解,得到淡黄色透明均相溶液;冷却至室温,注射加入CS-UCNPs(core-shell),加热至110℃,反应30 min以去除体系中的氯仿,降至室温后缓慢匀速加入含有1 mmol NH4F和0.63 mmol NaOH的5 mL甲醇溶液,剧烈搅拌,形成白色浑浊液.后续步骤与之前相同,最后得到核/壳/壳UCNPs(CSS-UCNPs)并分散在4 mL三氯甲烷中,置于4℃冰箱中待用.
在文献[29]报道的高温共沉淀法的基础上进行部分改进,合成核发光UCNPs(C-UCNPs)的核层.向100 mL三颈烧瓶中加入n(Y)/n(Yb)/n(Er)=0.80/0.18/0.02的0.75 mmol稀土盐混合物、11.5 mL十八烯和4.5 mL油酸,其它合成具体步骤与之前相同.
1.2.2 配体交换法合成PEI修饰的UCNPs采用配体交换法对C-UCNPs和CSS-UCNPs进行PEI修饰,合成了一系列PEI-UCNPs.首先,称取300 mg PEI置于50 mL三口烧瓶中,加入20 mL一缩二乙二醇,常温下搅拌至溶液均相澄清,抽真空30 min后,在氩气保护下升温至110°C;随后,向体系内注射加入4 mL UCNPs,反应45 min以除去溶液中的氯仿;升温至240°C并搅拌反应5 h,加入20 mL无水乙醇使沉淀析出,用无水乙醇洗涤沉淀,以13000 r/min转速离心5 min,重复此操作4次,最后超声分散至3 mL超纯水中.
1.2.3 金纳米颗粒(AuNPs)和DNA功能化金纳米颗粒(DNA-AuNPs)的制备参照文献[30]方法并稍作改进,采用柠檬酸还原法制备13 nm AuNPs.取1.03 mL浓度为25.4 mmol/L的HAuCl4水溶液置于100 mL烧瓶中,加入23.97 mL超纯水,水浴加热至130℃,持续沸腾15 min,随后加入2.5 mL柠檬酸钠(38.8 mmol/L),溶液逐渐变为酒红色,继续加热15 min后搅拌,冷却至室温,所得溶液避光置于4℃冰箱中储存备用.
采用经典盐化法制备DNA-AuNPs[31].分别取120 μL DNA 1(20 μmol/L)和120 μL DNA 2(与TCEP在室温孵育1 h)加入到800 μL AuNPs原液中,室温下孵育24 h后,每隔20 min缓慢加入5 μL 2 mol/L NaCl溶液至NaCl终浓度为0.2 mol/L;室温下孵育16 h后,用10 mmol/L的T-OAC缓冲液洗涤5次以除去剩余未反应的DNA分子,最后分散至400 μL T-OAC缓冲液中,避光置于4℃冰箱中储存备用.
1.2.4 HPV16 DNA的双信号检测HPV16 DNA的双信号检测分为比色检测和荧光检测.取0.6 mL无酶离心管分别加入2 μL LbCas12a蛋白(1 μmol/L)、3 μL CrRNA(1 μmol/L)和43.5 μL NEB CutSmart Buffer 2.1缓冲溶液,充分混匀,置于30℃摇床上振荡孵育30 min,分别加入1 μL不同浓度的目标双链DNA与1.5 μL Linker DNA(10 μmol/L),于30℃摇床上振荡孵育2 h,再分别加入50 μL DNA 1-AuNPs和DNA 2-AuNPs,于30℃摇床上振荡孵育30 min后,通过肉眼进行比色;随后分别加入20 μL预先制备的氨基功能化的CSS-UCNPs,于30℃摇床上振荡孵育1 h后,用配有980 nm激光器的荧光仪测量上转换荧光强度.
2 结果与讨论
2.1 上转换发光纳米材料的表征

Fig.1 Powder XRD patterns of C-UCNPs(A)and CSS-UCNPs(B)
NaYF4基质有2种常见晶型,一种为立方相(α),另一种为六方相(β).据文献[32]报道,六方相NaYF4的晶体结构对称性较低,而对称性低的晶体场有利于发光离子的f-f跃迁,与立方相NaYF4相比,六方相NaYF4的发光效率可提高约1个数量级.因此,为了获得高发光效率的UCNPs,首先利用高温共沉淀法合成了2种不同结构的六方相UCNPs(C-UCNPs和CSS-UCNPs),然后对这些UCNPs进行了XRD测试,用于确定基质晶型结构.从粉末晶体衍射结果(图1)可以看出,2种UCNPs晶型谱图的晶体衍射峰与六方相NaYF4材料(JCPDs:28-1192)标准卡片的衍射峰完全一致,表明已合成纯六方相基质的2种UCNPs.
为了分析制备的C-UCNPs和CSS-UCNPs的形貌和尺寸,进行了TEM表征.从图2(A)和图3(A)~(C)可以看出,2种UCNPs均呈均匀的圆棒形.由图3(A)可知,CSS-UCNPs的核C1-UCNPs呈圆棒形,轴向粒径约为29.10 nm,径向粒径约为21.75 nm[图3(G)和(J)];然后通过高温共沉淀、介导壳层外延成长法,以C1-UCNPs为种子,在其表面生长一层发光壳层,制得CS-UCNPs.如图3(B)所示,CSUCNPs呈圆棒形,轴向粒径约为38.91 nm,径向粒径约为22.85 nm[图3(H)和(K)].与C1-UCNPs比较可知,发光壳层主要生长在轴向,在轴向约生长4.9 nm;最后通过同样的方法,以CS-UCNPs为种子,在CS-UCNPs表面生长一层惰性壳层,制得CSS-UCNPs.由图3(C)可知,CSS-UCNPs呈圆棒形,轴向粒径约为43.26 nm,径向粒径约为24.28 nm[图3(I)和(L)].与CS-UCNPs比较可知,惰性壳层也主要生长在轴向,轴向和径向生长的差异性可能跟晶体的异向生长有关.HRTEM[图2(B)和图3(D)~(F)]表明,C-UCNPs,C1-UCNPs,CS-UCNPs和CSS-UCNPs具有较清晰的晶格条纹,晶面间距约为0.52 nm,与六方相NaYF4的(100)晶面参数一致.通过HRTEM和XRD分析,进一步证明了合成的2种UCNPs均为六方晶相.

Fig.2 TEM(A)and HRTEM(B)images,axial(C)and radial(D)particle size statistics of C-UCNPs
为了获得材料表面分子的官能团或化学键等信息,对制备的UCNPs进行了红外光谱(FTIR)表征,验证了UCNPs表面油酸分子的存在.如图4所示,在C-UCNPs和CSS-UCNPs的红外光谱图中均检测到1557和1464 cm-1处的2个邻近吸收峰,归属于羧酸根离子的对称和反对称振动吸收峰.综上,确定合成了油酸包覆的UCNPs.
为了进一步验证C-UCNPS和CSS-UCNPS的元素组成,进行了能谱(EDS)表征.从C-UCNPS的EDS能谱图[图5(A)]可知,敏化剂Yb离子和激活剂Er离子均已掺杂到NaYF4基质中,Na,F,Y,Er和Yb的原子百分比分别为14.36%,70.91%,12.89%,0.12%和1.72%,从C-UCNPs的mapping元素图(图6)可知,Na,F,Y,Yb和Er元素均分布在C-UCNPs上.从CSS-UCNPS的EDS能谱图[图5(B)]可知,敏化剂Yb离子和激活剂Er离子均已掺杂到CS-UCNPs和CSS-UCNPs中,CS-UCNPs的Na,F,Y,Er和Yb的原子百分比分别为13.48%,74.85%,10.64%,0.10%和0.92%,CSS-UCNPs的Na,F,Y,Er和Yb的原子百分比分别为14.85%,70.97%,12.90%,0.41%和0.87%,从图7的mapping元素图中可看出,Na,F和Y元素均分布在C1-UCNPs上[图7(A)],Na,F,Y,Er和Yb元素均分布在CS-UCNPs[图7(B)]和CSS-UCNPs[图7(C)]上.
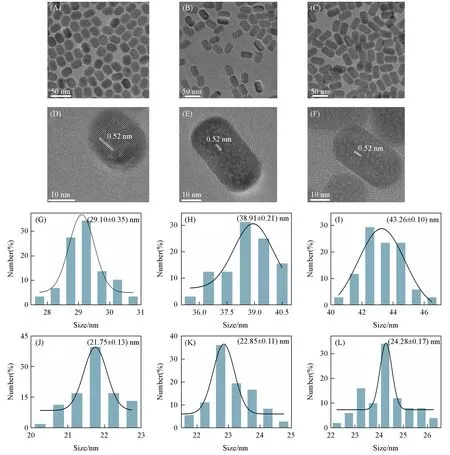
Fig.3 TEM(A—C)and HRTEM(D—F)images of C1-UCNPs(A,D),CS-UCNPs(B,E)and CSS-UCNPs(C,F),axial particle size statistics(G—I)and radial particle size statistics(J—L)of C1-UCNPs(G,J),CS-UCNPs(H,K)and CSS-UCNPs(I,L)
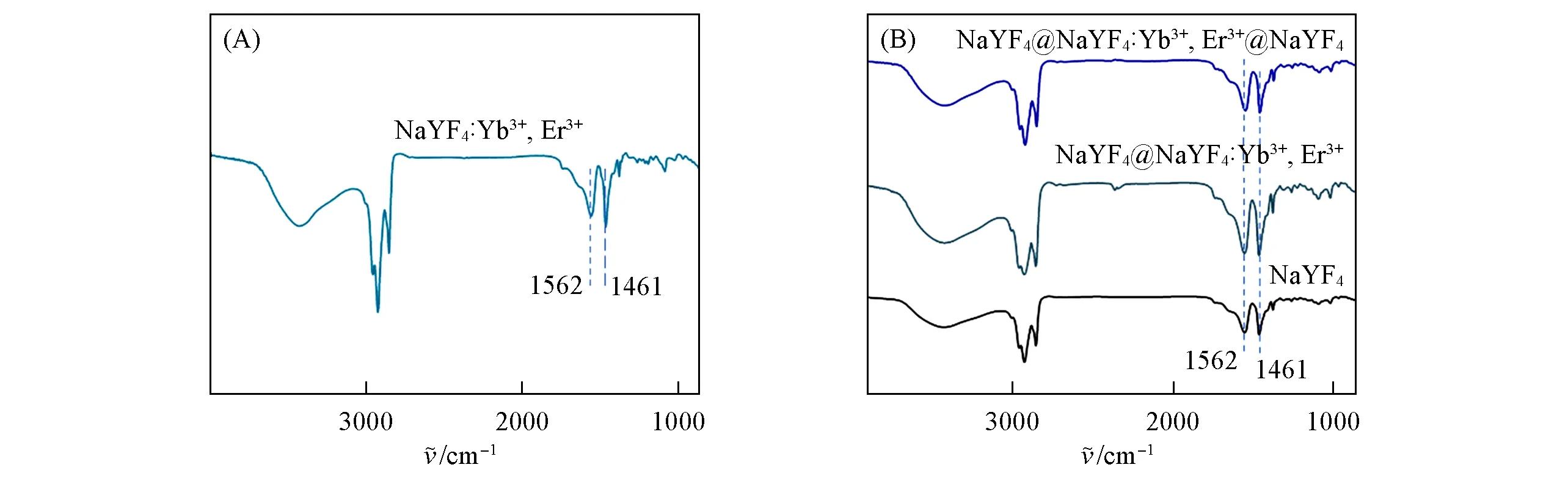
Fig.4 FTIR spectra of C-UCNPs(A)and CSS-UCNPs((B)
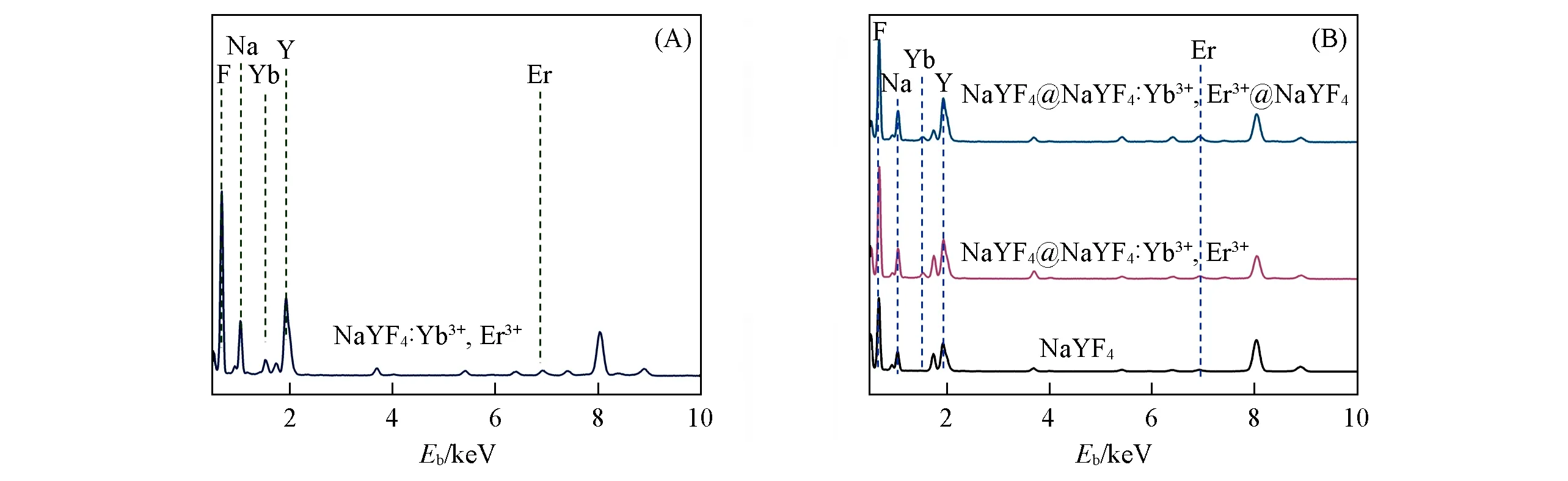
Fig.5 EDS spectra of C-UCNPs(A)and CSS-UCNPs(B)
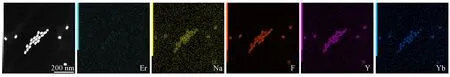
Fig.6 EDS element mappings of C-UCNPs

Fig.7 EDS element mappings of C1-UCNPs(A),CS-UCNPs(B)and CSS-UCNPs(C)
2.2 表面修饰PEI分子的UCNPs的表征
PEI分子中含有氨基基团,带正电.当UCNPs表面修饰上PEI分子后表面配体和表面电势会发生改变,因此利用FTIR和动态光散射(DLS)分别对合成的UCNPs进行表征,以判断UCNPs表面是否修饰了亲水性的PEI配体.未修饰PEI的UCNPs检测到1562和1461 cm-1处的2个邻近吸收峰[图8(A)],归属于羧酸根离子的对称和反对称振动吸收峰;zeta电势图表明未修饰PEI的UCNPs带负电[图8(B)],这是因为未修饰PEI分子的UCNPs表面配体是油酸分子,含有羧基,带负电;修饰PEI分子后,在1637 cm-1处出现了吸收峰[图8(A)],归属于氨基的弯曲振动,zeta电势图表明亲水UCNPs明显带正电[图8(B)].结合上述分析可知,在UCNPs表面已修饰上了PEI分子.

Fig.8 FTIR spectra(A)and DLS zeta potential(B)of UCNPs
2.3 金纳米颗粒(AuNPs)和DNA功能化金纳米颗粒(DNA-AuNPs)的表征
采用文献[30]报道的柠檬酸钠还原法合成AuNPs,并通过TEM和紫外-可见分光光度计对其进行表征.如图9(A)所示,制备的AuNPs尺寸较均一,分散性良好;粒子直径约为12.9 nm[图9(B)];如图9(C)所示,AuNPs在521 nm处吸收峰强度最大,证明已合成了13 nm的AuNPs,可用于后续实验.

Fig.9 TEM image of AuNPs(A),particle size statistics of AuNPs(B),UV-Vis absorption spectra of AuNPs with different concentrations(C),standard curve of AuNPs absorbance and concentration(D),UV-Vis absorption spectra of AuNPs and DNA-AuNPs(E)and zeta potential diagram of AuNPs and DNA-AuNPs(F)
采用盐化法实现了巯基DNA与AuNPs的共价偶联,在AuNPs和巯基DNA偶联过程中,DNA和AuNPs的浓度比对偶联效率有较大影响,因此需要通过建立吸光度值(A)与AuNPs浓度之间的标准曲线获得AuNPs的浓度.如图9(D)所示,将521 nm处的吸光度A对一系列已知的AuNPs浓度进行线性拟合,得到吸光度值A与AuNPs浓度的线性方程:A=0.2864cAuNPs-0.0841[图9(B)],通过代入该方程可计算出AuNPs的浓度;随后控制ssDNA浓度,确保AuNPs与DNA的偶联效率.参照文献[33]报道的DNA和AuNPs浓度比并对其稍做调整制备了DNA-AuNPs,当DNA修饰在AuNPs上后,AuNPs的紫外吸收峰出现红移.此外,由于DNA分子含有磷酸基团使其带有负电,与AuNPs共价键连接后,AuNPs的电势会进一步降低.基于这些性质,对DNA-AuNPs进行了紫外-可见吸收光谱及DLS电势测试.如图9(E)所示,与DNA偶联后AuNPs的最大吸收波长发生红移,由521 nm移动到527 nm;DLS电势测试结果表明,与AuNPs相比,DNA-AuNPs电势明显变负[图9(F)].以上结果均与预期一致,表明制备了DNAAuNPs.
2.4 可行性分析
由图10(A)可知,AuNPs的UV-Vis吸收光谱与UCNPs在542 nm处的发射峰有较大重叠;由图10(B)可知,PEI-UCNPs带正电,AuNPs带负电,二者可通过静电作用吸附到一起.综上可知,当PEI-UCNPs作为能量供体,AuNPs作为能量受体时,两者满足发光共振能量转移条件,AuNPs可以有效猝灭PEI-UCNPs在542 nm处的绿光发射.

Fig.10 UV-Vis absorption spectra of AuNPs and luminescence spectra of UCNPs(A)and zeta potential diagram of AuNPs and UCNPs(B)
2.5 两种发光结构上转换纳米材料的比较
UCNPs修饰PEI配体的过程中,由于UCNPs会与环境中的物质发生能量交换产生表面猝灭效应而引起上转换纳米材料发光强度的变化,且不同结构的UCNPs发光强度变化情况有所不同,为此分别测定了2种不同结构UCNPs修饰PEI配体前后的发光光谱.由图11可知,CSS-UCNPs的发光强度变化最小,说明通过惰性壳层包覆的策略可以有效避免表面猝灭效应的产生,提高抗外界环境干扰的能力.
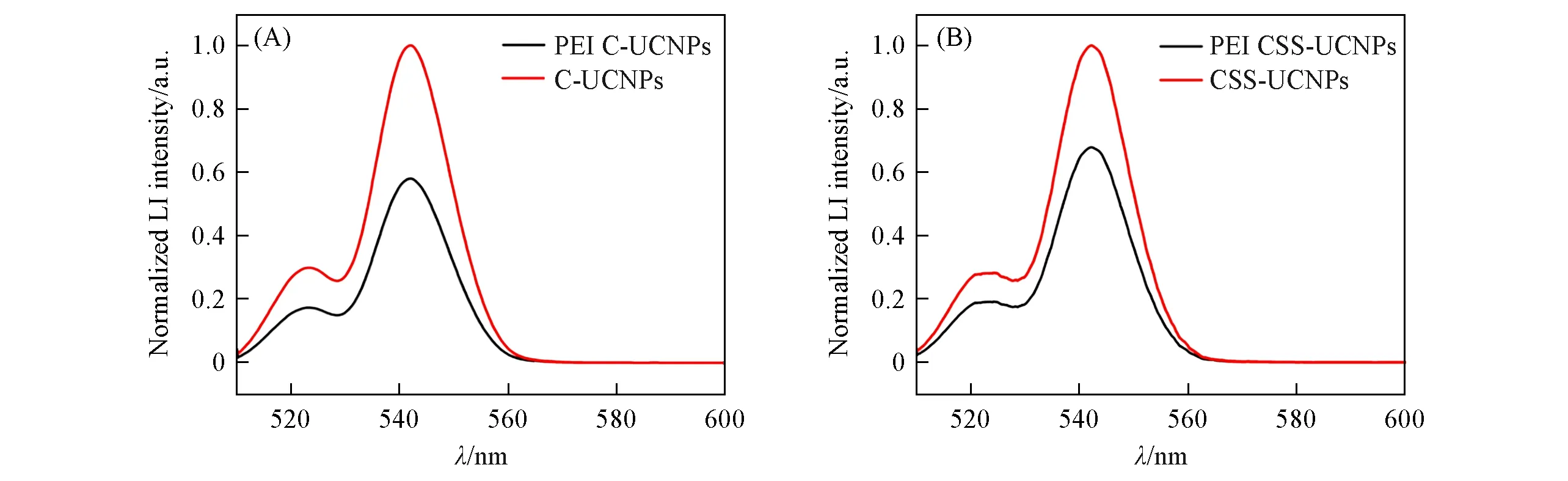
Fig.11 Luminescence spectra of PEI modified C-UCNPs(A)and CSS-UCNPs(B)
实验中探究了不同结构上转换纳米材料与DNA-AuNPs之间的LRET效率.从图12(A)和(B)的TEM照片可看出,DNA-AuNPs较为均匀地分布在PEI-UCNPs上,表明在静电作用力下,DNA-AuNPs可吸附在PEI-UCNPs周围,进一步证明了本方法的可行性.从图12(C)和(D)可知,2种PEI UCNPs的猝灭效率分别为57.9%和77.5%,PEI C-UCNPs的猝灭效率较低,这是因为发光裸核是一个几十纳米的实心发光圆棒体,实现大部分猝灭实心发光体的发光难度较大,而CSS-UCNPs为几纳米的发光层,因此PEI CSS-UCNPs猝灭程度比较高.综上分析,最终选择了PEI CSS-UCNPs.

Fig.12 TEM images(A,B)and LRET spectra(C,D)of PEI C-UCNPs,PEI CSS-UCNPs with DNA-AuNPs
2.6 双信号检测和特异性验证
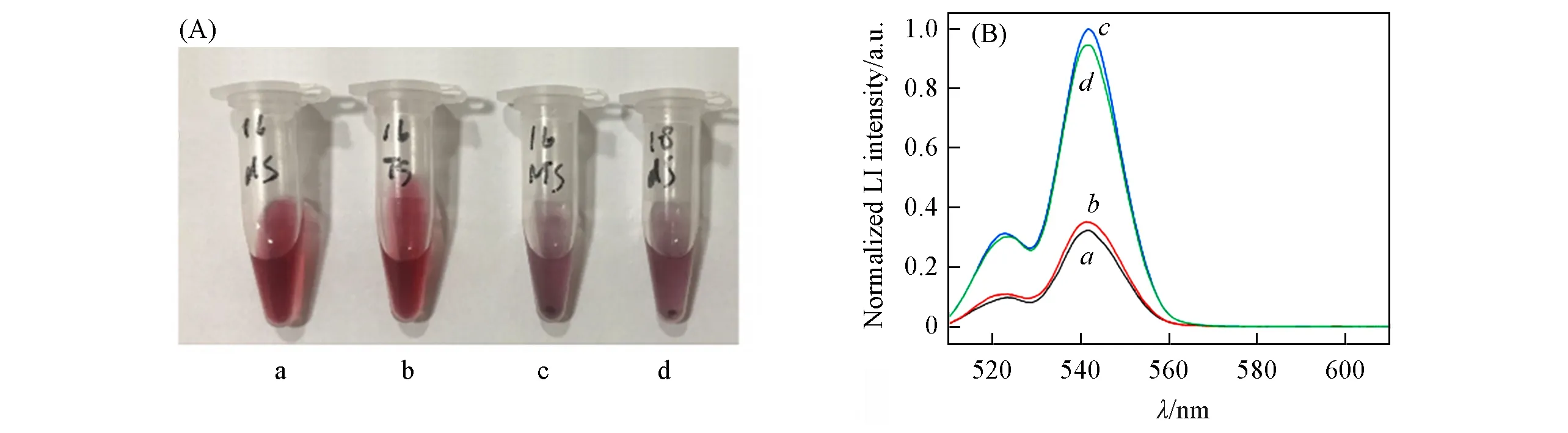
Fig.13 Photograph of colorimetric(A)and luminescence spectra(B)of this biosensor in the presence of HPV16 dsDNA(a,a),HPV16 TS DNA(b,b),HPV16 NTS DNA(c,c)and HPV18 dsDNA(d,d)
CRISPR-Cas12a(Cpf1)蛋白是RNA引导的酶,作为细菌适应性免疫系统的组成部分可结合和切割DNA.CRISPR/Cas12a蛋白的切割活性有2种激活方式:第一种是在CrRNA的作用下可以特异性靶向识别包含原型间隔序列(PAM)的目标双链DNA(dsDNA),在顺式切割序列特异性的目标dsDNA之后,Cas12a蛋白对于单链DNA(ssDNA)的反式切割活性也被激活,可以无差别地切割其附近的任何ss DNA;第二种是在CrRNA的作用下可以特异性靶向识别不具有PAM序列的ssDNA,激活反式切割活性,非特异性切割任何非目标ssDNA.本实验选择第一种激活方式,使用含有PAM序列的HPV16 dsDNA,激活CRISPR/Cas12a的切割活性,实现对Linker ssDNA的切割降解,从而通过体系颜色和光学信号的双信号改变实现对目标物的检测.为了验证这一核心观点,首先使用HPV16 dsDNA,HPV18 dsDNA,HPV16 TS ssDNA和HPV16 NTS ssDNA作为检测物,验证CRISPR-Cas12a反式切割Linker ss DNA的活性.从图13(A)中的颜色变化可以看出,CrRNA可特异性识别并结合HPV16 dsDNA和HPV16 TS ssDNA,进而激活CRISPR/Cas12a对Linker ssDNA的切割活性,这与CRISPR-Cas12a系统两种激活方式相对应.由于Linker ssDNA断裂,DNA-1/2-AuNPs无法发生连接,保持分散状态,溶液呈现红色;同时,CrRNA无法识别结合HPV18 dsDNA和HPV16 NTS ssDNA,不能激活CRISPR/Cas12a的切割活性,Linker ssDNA保持完整,DNA-1/2-AuNPs稳定连接,使得AuNPs发生团聚,溶液变为蓝紫色.加入PEI CSS-UCNPs与上述体系混匀孵育1 h后,采集上转换发光信号,从图13(B)可知,团聚程度不同的DNA-AuNPs对UCNPs在542 nm处的猝灭效果不同,分散状态的AuNPs可明显猝灭上转换在542 nm处的发光.另外,相较于不具有PAM序列的ssDNA,具有PAM序列的dsDNA与CRISPR/Cas12a结合后激活的DNA切割活性更高,且dsDNA的NTS链有助于将Cas12a复合物稳定在最佳构象,进而实现对ssDNA的反式切割[28].因此最终选择HPV16 dsDNA作为目标物激活CRISPR/Cas12a的反式切割活性从而实现对Linker ssDNA的切割降解,产生上转换发光信号的改变实现对目标物的检测.
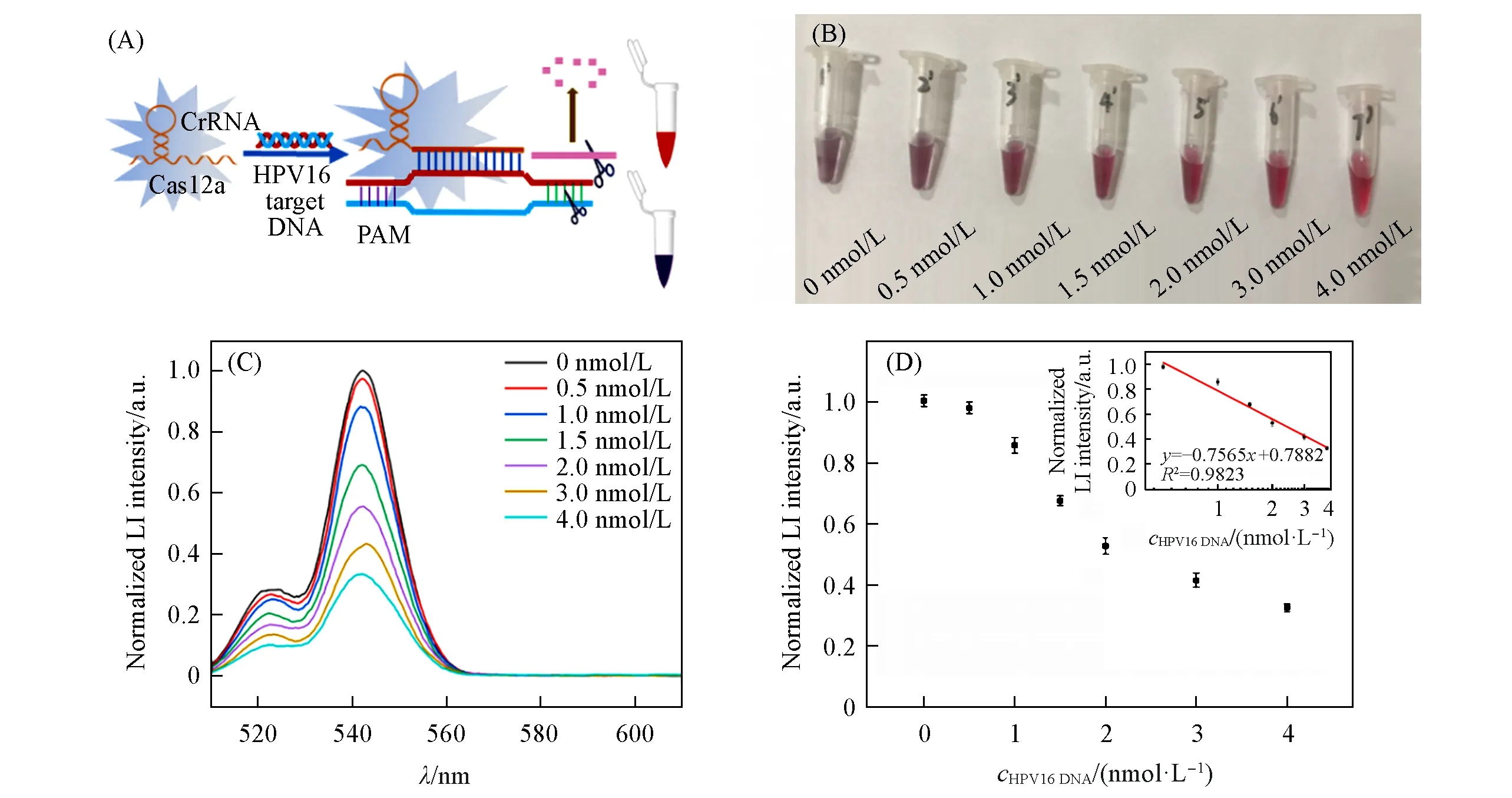
Fig.14 Schematic diagram of colorimetric detection principle(A),photograph of colorimetric detection of different concentrations of HPV16 DNA(B),luminescence spectra of different concentrations of HPV16 DNA(C)and scatter diagram of luminescence with different concentrations of HPV16 DNA(D)The inset in(D)is the working curve between the logarithm of HPV16 DNA and normalized luminescence intensity.
由上述结果可知,HPV16 dsDNA可以激活CRISPR/Cas12a的切割活性进而实现对Linker ssDNA的切割降解,Linker ssDNA的完整性会影响DNA-AuNPs间的团聚状态[图14(A)];通过检测不同DNA-AuNPs间团聚状态下的PEI CSS-UCNPs的发光信号,可建立发光信号与不同HPV 16 dsDNA浓度的关系,实现对HPV16 DNA的检测.对实验条件进行优化(图S1和图S2,见本文支持信息)后,选择100 nmol/L Linker DNA,30℃,T-OAC反应缓冲液作为连接2种DNA-AuNPs的实验条件;选择NEB CutSmart Buffer 2.1反应缓冲溶液,Cas12a∶CrRNA浓度比为1∶1.5,反应时间2 h,反应温度30℃作为激活CRISPR-Cas12a系统切割能力的实验条件.在上述反应条件下,检测了不同浓度的HPV16 dsDNA(0,0.5,1,1.5,2,3和4 nmol/L).本文方法分为比色和上转换双信号检测,首先可以通过观察颜色初步判断是否存在目标物.手机拍照后,加入PEI CSS-UCNPs混匀孵育1 h,采集上转换发光信号进行定量检测,进一步提高检测准确性和灵敏度.从图14(B)中的颜色变化可以看出,当HPV16 DNA的浓度从0向4 nmol/L逐渐增加时,溶液颜色发生明显变化,由蓝紫色逐渐向紫红色过渡,直至最后变成红色,初步完成目标物的定性检测.随后,为了对HPV16 DNA进行定量检测,绘制了以上溶液的上转换发光谱图[图14(C)],并用UCNPs 542 nm处的发光强度和目标物浓度绘制标准曲线[图14(D)],对其进行线性拟合.如图14(E)所示,HPV16 dsDNA浓度的对数值与上转换发光强度线性相关,检测范围为0.5~4 nmol/L,根据检出限的定义3σ/k(σ为空白样品的标准偏差,k为工作曲线的斜率),可计算得出发光法的检出限为69.8 pmol/L.
为了验证该双信号检测方法对目标物HPV16 DNA片段识别的特异性,选取了分别与目标HPV16 DNA单碱基错配、双碱基错配、三碱基错配的DNA序列、HPV18 DNA和空白对照,所有DNA序列的浓度均为4 nmol/L,按照上述流程对这几种DNA序列进行比色和发光检测.如图15(A)所示,HPV16 DNA和单碱基错配分别呈现红色和暗红色,其余DNA片段均呈现蓝紫色,表明特异性较好,通过肉眼可明显区分目标DNA和单碱基错配的目标DNA,可完成初步定性检测;随后加入UCNPs后采集发光信号绘制柱状图,如图15(B)所示,HPV16 DNA的上转换发光在542 nm处的发光强度最低,单碱基错配的次之,其它DNA片段的发光强度均比较高.综合上述分析可知,建立的HPV16 DNA双信号检测方法具有良好的特异性,并且可以实现单碱基错配检测,提高了DNA片段的容错率.

Fig.15 Photographs of colorimetric detection of different DNA molecules(A)and specificity investigation toward different DNA molecules(B)a.HPV16;b.MT1;c.MT2;d.MT3;e.HPV18;f.blank.
3 结论
通过多步高温共沉淀法合成了C-UCNPs和CSS-UCNPs,并对这些UCNPs的晶型、形貌、表面配体和发光共振能量转移效率进行了表征.结果表明,制备了表面猝灭效应低、发光共振转移效率较高的CSS-UCNPs.随后,制备了形貌较为均匀的DNA功能化的AuNPs(DNA-AuNPs).将氨基修饰的CSSUCNPs与CRISPR/Cas12a-AuNPs复合系统结合,基于AuNPs比色和上转换发光双信号灵敏检测HPV16 DNA.通过肉眼观察不同团聚程度AuNPs的颜色变化,可以实现对目标物的初步即时检测;又利用UCNPs发光信号的输出,进一步实现对目标DNA的定量检测,检出限为69.8 pmol/L,这说明双信号检测有效地提高了检测结果的准确性.此外,该方法不仅特异性强,而且还能识别单碱基错配的HPV16 DNA,提高了DNA片段的容错率.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/20220412.