辛基羟肟酸在氟碳钙铈矿表面吸附的密度泛函理论研究
2022-11-08王介良程泽宇
王介良 刘 龙 程泽宇 曹 钊
(1.内蒙古科技大学矿业与煤炭学院,内蒙古 包头 014010;2.内蒙古矿业工程重点实验室,内蒙古 包头 014010;3.白云鄂博共伴生矿资源高效综合利用省部共建协同创新中心,内蒙古 包头 014010)
稀土是极重要的战略金属资源,目前已知稀土矿物大约有169种,具有工业开采和实用价值的稀土矿物仅十几种,主要包括氟碳铈矿、独居石、氟碳钙铈矿、铈铌钙钛矿、磷钇矿、褐钇铌矿、离子吸附型稀土矿等[1-2]。其中氟碳钙铈矿常与氟碳铈矿伴生,在我国白云鄂博矿、四川牦牛坪稀土矿及微山湖稀土矿均有分布[3],美国雪鸟稀土矿[4]、哥伦比亚穆佐翡翠稀土矿[5]则是以氟碳钙铈矿为主的稀土矿。
浮选是回收稀土的主要方法,矿物晶体化学性质影响浮选药剂在矿物表面的吸附,是决定稀土与脉石矿物可浮性差异的根本原因[6]。目前,稀土矿选矿技术及理论研究主要集中于氟碳铈矿、独居石等稀土矿物[7-8]。CAO 等[9]采用密度泛函理论计算,研究了氟碳铈矿结构和电子性质及表面性质对浮选的影响机制;ESPIRITU 等[10]通过浮选、第一性原理计算等方法,研究了矿物溶解组分对氟碳铈矿、独居石和白云石浮选的影响;史新章等[11]采用密度泛函理论计算,发现辛基羟肟酸(OHA)通过取代水,能在独居石表面形成五元环吸附态。而针对氟碳钙铈矿的研究甚少,尤其在氟碳钙铈矿晶体化学性质方面的研究十分欠缺。
本文基于密度泛函理论,采用分子模拟方法,研究氟碳钙铈矿解离面特性、能带结构、态密度、电荷密度及差分电荷密度等表面性质,探明阴离子捕收剂辛基羟肟酸在氟碳钙铈矿表面的吸附机制,为氟碳钙铈矿选矿实践提供理论依据,完善稀土氟碳酸盐矿物浮选理论体系。
1 计算及试验方法
氟碳钙铈矿DFT 计算参数设置:采用Material Studio 2017 软件中的CASTEP 模块,在密度泛函理论原则下,对氟碳钙铈矿(100)面优化、OHA 分子优化和药剂吸附计算,选取GGA-PBESol 作为交换关联参数,平面波截断能设置为450 eV,k 点取样密度为3×3×1,体系选用OTFG 超软赝势[12],smearing 设置为0.1。几何优化收敛标准设定如下:最大能量收敛极限1×10-5eV/atom,原子间作用力收敛标准0.3 eV/nm,原子间最大内应力0.05 GPa,原子最大位移1×10-4nm,设定自洽迭代收敛标准为1×10-5eV/atom。对优化后的晶胞模型切分,构建表面模型后,对晶面真空层厚度及原子层数进行收敛性测试,以确定最稳定的表面模型,其真空层厚度为1 nm;表面能带、态密度及电荷密度差分性质计算,收敛精度选取1×10-6eV,其余参数保持参数一致,所有计算均在倒易空间中进行。
分别采用式(1)和式(2)对氟碳钙铈矿不同晶面的断裂键密度(Db)以及药剂在选定解离面吸附能(ΔE)进行计算:
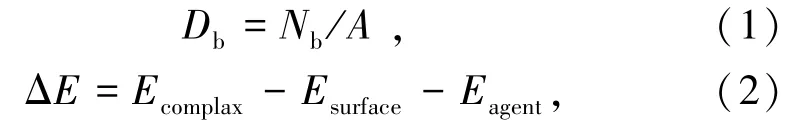
式中:Db、Nb和A分别为晶面断裂键密度、单位晶面断裂键个数和解离面面积;ΔE表示吸附能;Ecomplax、Esurface和Eagent分别表示矿物表面吸附络合物、矿物表面和药剂的总能量。吸附能为负值时表明吸附过程可自发进行,吸附能越小,代表着吸附越牢固;吸附能为零或正值时表明吸附过程不能自发进行。
2 试验结果与讨论
2.1 表面结构弛豫
氟碳钙铈矿XRD 和单胞模型分别如图1(a)和图1(b)所示。由图1(a)可知,氟碳钙铈矿常见暴露面为(119)、(100)及(110)面。氟碳钙铈矿晶胞模型基于Ni[13-14]研究建立,氟碳钙铈矿为单斜晶系,Cc空间群,晶胞参数为a=1.231 nm,b=0.711 nm,c=2.825 nm;α=90°,β=98.24°,γ=90°。晶胞由2个Ca 层,4个(CeF)层以及6个(CO3)层组成,其中Ca为8 配位,连邻近两层各4个O 原子;Ce 原子连接上下两层各3个O 原子及同层3个F 原子。其中C—O键为共价键,其余均以离子键相连。
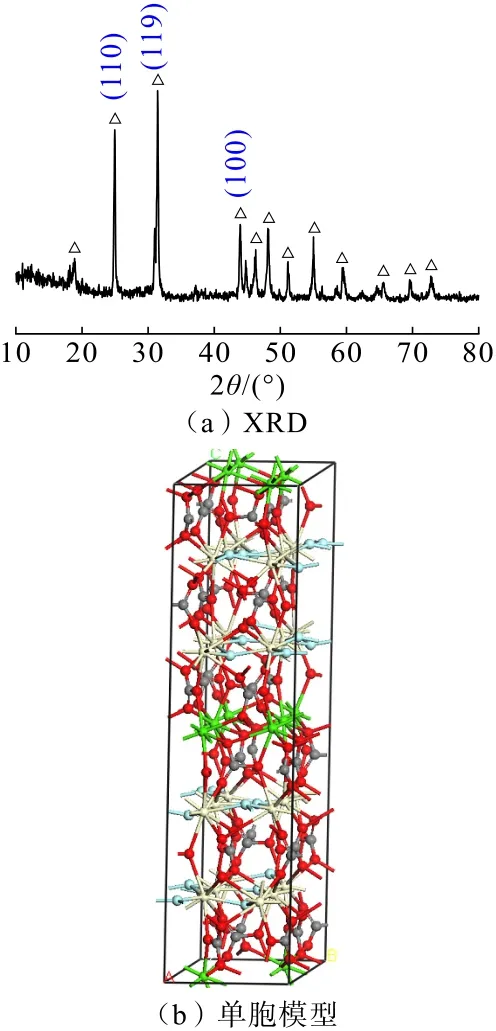
图1 氟碳钙铈矿XRD 和单胞模型Fig.1 XRD spectra and unit cell model for parisite
采用MS 软件中Build Surface 模块分别构建氟碳钙铈矿(119)、(100)及(110)面并进行优化,进行各解离面断键密度与表面能计算,结果如表1所示。由表1 可知,氟碳钙铈矿(119)、(100)及(110)面断键密度分别为19.82 nm-2、13.44 nm-2和16.12 nm-2,(100)与(110) 面表面能分别为0.67 J/m2、1.14 J/m2,而(119)面断裂键密度最高,且观察中发现,更换不同Top 值形成的表面,始终无法形成完整的CO32-基团,而CO32-基团中,C—O 键共价作用强,表面解理过程中,很难发生断裂,故弃用该表面,由此可知氟碳钙铈矿(100)表面断裂键密度最低,表面能最小,在矿物粉碎过程中优先解离。
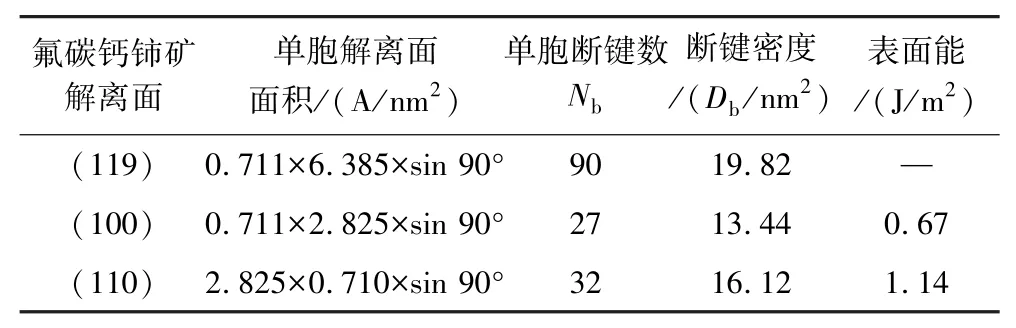
表1 氟碳钙铈矿常见解离面断键密度及表面能计算Table 1 Calculation of broken bonds density and surface energy of commonly cleavage planes on parisite
对氟碳钙铈矿(100)面进行表面弛豫,弛豫结果如表2 和图2所示。由表2 和图2 可知,氟碳钙铈矿表层原子均产生弛豫现象,x轴方向的弛豫最为明显,z轴方向次之,y轴方向的弛豫较弱。其中,Ca 1、Ce 3、Ce 5 原子向体相内部弛豫,Ce 1 向解离面外部弛豫。O、F 原子多向解离面外部弛豫。弛豫结果表明氟碳钙铈矿(100)面整体弛豫小,没有明显的表面重构现象产生。

图2 氟碳钙铈矿(100)解离面原子弛豫前后的表面结构Fig.2 Surface structure of parisite (100) cleavage surface before and after atoms relaxation

表2 氟碳钙铈矿(100)面表面原子位移Table 2 The atomic displacements of parisite (100) surface
2.2 能带结构分析
氟碳钙铈矿体相和(100)面能带结构如图3所示。由图3(a)可知,以费米能级(EF)为能量零点,氟碳钙铈矿在费米能级附近存在禁带,带宽小于5 eV,宽度较窄,且导带底部接近费米能级,结果表明氟碳钙铈矿为半导体。由图3(b)可知,氟碳钙铈矿(100)面的价带较氟碳钙铈矿体相向上移动,靠近费米能级处的价带新增多个能级,价带顶位于-2 eV,表明表面的出现有可能使氟碳钙铈矿原价带能级发生分裂。禁带宽度是半导体的一个重要特征量,反映的是晶体表面电子的束缚强度,其大小与晶胞结构和晶胞内原子间的结合性质有关,禁带宽度越小,电子越容易吸收较小的能量而跃迁到导带,产生本征激发[16]。由于氟碳钙铈矿(100)面价带上移,禁带宽度变窄,产生本征激发的能量减小,电子活性增强,使表面原子更容易转移电子发生反应;而(100)面导带下移,与费米能级相连,(100)面金属特征增强,得电子能力增强,浮选时更容易与阴离子药剂作用而产生吸附。

图3 氟碳钙铈矿体相和氟碳钙铈矿(100)面能带结构Fig.3 Energy bands of parisite bulk and parisite (100) surface
2.3 态密度分析
态密度反映电子在特定能级的分布情况,一般来说,处于低能级的电子相对稳定,处于高能级的电子不稳定,活性较强[17-18]。氟碳钙铈矿体相的总态密度图和局域分波态密度图如图4所示。由图4 可知,氟碳钙铈矿态密度在-45~30 eV 之间分布,-45~30 eV 之间,主要由Ce 和Ca 的s 轨道参与贡献;-30~15 eV 之间,Ce、Ca、F、O 和C 原子轨道都有贡献;-15~0 eV 之间,主要由O 和C 原子的s、p 轨道,F 原子的p 轨道,以及Ca 原子的s、d 轨道参与贡献;0~25 eV之间,主要由Ce原子的d、f轨道,Ca和C原子的所有轨道,以及F 和C 原子的p 轨道参与贡献,Ce 原子的f 轨道做主要贡献。Ce 原子的f 轨道对费米能级附近的态密度贡献最大,Ca 原子轨道次之。研究表明,费米能级附近的原子电子活性最强[19-20],因此可知氟碳钙铈矿中Ce 原子的活性较强,其次是Ca原子,在浮选过程中易与浮选药剂发生化学反应。

图4 氟碳钙铈矿态密度Fig.4 State density of parisite
氟碳钙铈矿体相与氟碳钙铈矿(100)面的Ce 原子和Ca 原子态密度分布如图5所示。由图5(a)可知,Ce 原子在-40~15 eV 区域的态密度由3 部分组成,与氟碳钙铈矿体相中Ce 原子态密度相比,(100)面Ce 3 和Ce 5 原子在-36 eV 处的s 轨道和在-18 eV 的p 轨道态密度峰明显负移。此外,(100)面Ce原子位于费米能级(EF)附近的f与d 轨道的态密度,穿过费米能级正移,导电性增强,电化学活性也因此增强;位于4 eV 附近的d 轨道态密度峰发生断裂,态密度峰变宽,局域性减弱,电子离域性增强;同时,相较体相中的Ce 原子,(100)面Ce 原子的f、d 轨道态密度峰强减弱,电子分布减少,表明(100)面Ce 3、Ce 5 原子发生断键,得电子能力增强;计算结果表明氟碳钙铈矿(100)面解离后Ce 断键形成电子受体,活性增强,成为活性位点,在浮选过程中易与浮选药剂发生反应。由图5(b)可知,Ca 原子在-45~20 eV 区域的态密度包括三部分,与氟碳钙铈矿体相中Ca 原子态密度相比,表面位于-45~-40 eV 区域的s 轨道态密度变化不明显,-20 eV 附近d 轨道态密度有所增强,0~20 eV 区域内态密度向费米能级移动,电子活性增强,且该区域内各轨道态密度分布趋于均匀化,s、p、d 态密度交织,电子的非局域性增强,Ca 原子的得电子能力增强,成为活性位点。计算结果表明,浮选过程中,阴离子药剂容易在氟碳钙铈矿表面Ce和Ca 原子位点发生吸附。

图5 氟碳钙铈矿Ce 原子和Ca 原子态密度Fig.5 State density of Ce and Ca of parisite
2.4 差分电荷密度分析
为进一步研究氟碳钙铈矿(100)面电荷分布及转移过程,进行差分电荷密度计算。差分电荷密度能直观呈现原子间成键及电荷分布情况,反映浮选过程中矿物表面活性位点与浮选药剂的作用方式[21-22]。氟碳钙铈矿(100)面解理前后Ce 原子与Ca 原子的局部电荷密度及差分电荷密度分别如图6 和图7 所示。电荷密度图中,颜色深浅表示电子云密度的大小,颜色越深,电子云密度越大,原子间的成键作用越强,表明得到或者失去电子越多[23-24]。

图6 氟碳钙铈矿体相和(100)面Ce 原子电荷密度与差分电荷密度图Fig.6 Charge density and charge density differences of Ce in parisite and on parisite (100) surface
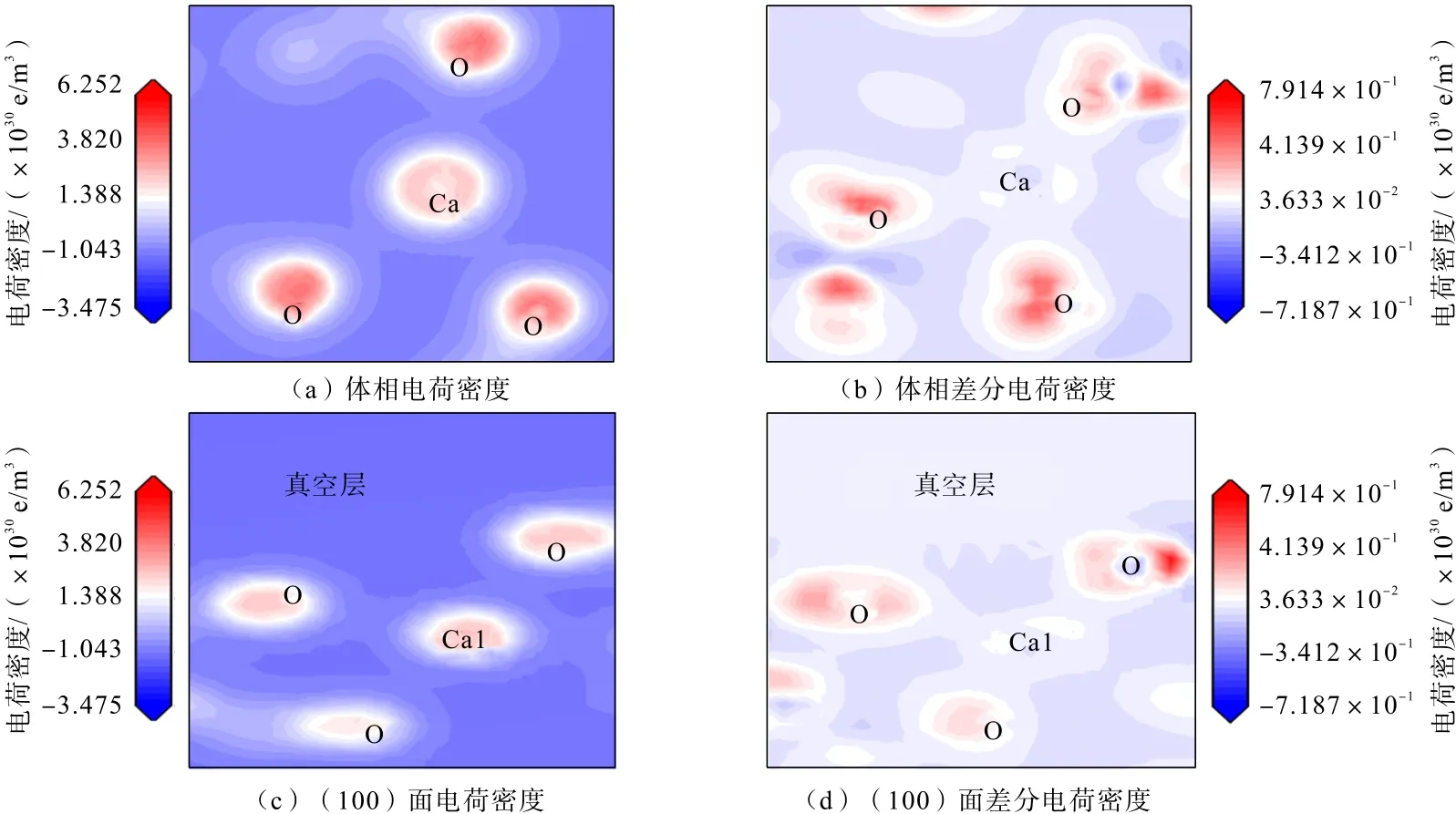
图7 氟碳钙铈矿体相和(100)面Ca 原子电荷密度与差分电荷密度图Fig.7 Charge density and charge density differences of Ca in parisite and on parisite (100) surface
图6(a)、(b)分别为氟碳钙铈矿体相Ce 和O 电荷密度及差分电荷密度图,图6(c)、(d)分别为氟碳钙铈矿(100)面Ce 和O 电荷密度和差分电荷密度图。
由图6(a)、(c)可知,氟碳钙铈矿(100)面Ce与O 原子之间的电荷密度高于氟碳钙铈矿体相,Ce与O 原子电荷密度重叠区域增强,氟碳钙铈矿(100)面中与Ce 原子相邻的O 原子的电荷密度低于氟碳钙铈矿体相;由图6(b)、(d)可知,氟碳钙铈矿(100)面与Ce 原子颜色较体相更深,表明Ce 原子失去电荷,O 原子得到电荷。计算表明氟碳钙铈矿(100)面Ce—O 键比体相中Ce—O 键键长更短,这是由于在形成(100)面过程中Ce 原子在表面产生断键,正电性增强,对O 原子吸引力增大,表面电子云重新分配所致。
图7(a)、(b)分别为氟碳钙铈矿体相Ca 和O 电荷密度及差分电荷密度图,图7(c)、(d)分别为氟碳钙铈矿(100)面Ca 和O 电荷密度和差分电荷密度图。
由图7(a)、(c)可知,氟碳钙铈矿(100)面Ca与O 原子之间的电荷密度高于氟碳钙铈矿体相,Ca与O 原子电荷密度重叠区域增强,氟碳钙铈矿(100)面中与Ca 原子相邻的O 原子的电荷密度低于氟碳钙铈矿体相;由图7(b)、(d)可知,(100)面中Ca 原子电荷密度变化不大,断键造成Ca 原子上方表面电子缺失,O 原子电荷密度颜色变浅,电子云往Ca 原子偏移,表面形成Ca 原子断键,Ca 原子正电性增强。
对比图6 和图7 可知,(100)面中Ce 原子正电性强于Ca 原子。在浮选中,带负电的阴离子药剂,能够与表面断键形成的带正电Ce 原子与Ca 原子作用,从而吸附在氟碳钙铈矿表面。
2.5 OHA 在氟碳钙铈矿表面吸附能计算
OHA 是稀土矿浮选研究常用的捕收剂,其在浮选溶液中的优势组分为OHA 阴离子[25-27],为判断OHA 阴离子在氟碳钙铈矿表面的吸附稳定性,对OHA 阴离子在氟碳钙铈矿(100)面Ce 原子和Ca 原子位点的单键吸附进行优化,图8所示为OHA 阴离子在氟碳钙铈矿表面Ce 原子吸附,OHA 在氟碳钙铈矿(100)面单核单配位、单核双配位、和双核双配位吸附前后的吸附构型,吸附能计算结果如表3所示。

表3 OHA 在氟碳钙铈矿(100)面Ce 原子和Ca 原子位点吸附能Table 3 Adsorption energy of OHA at Ce and Ca sites on (100) surface of parisite
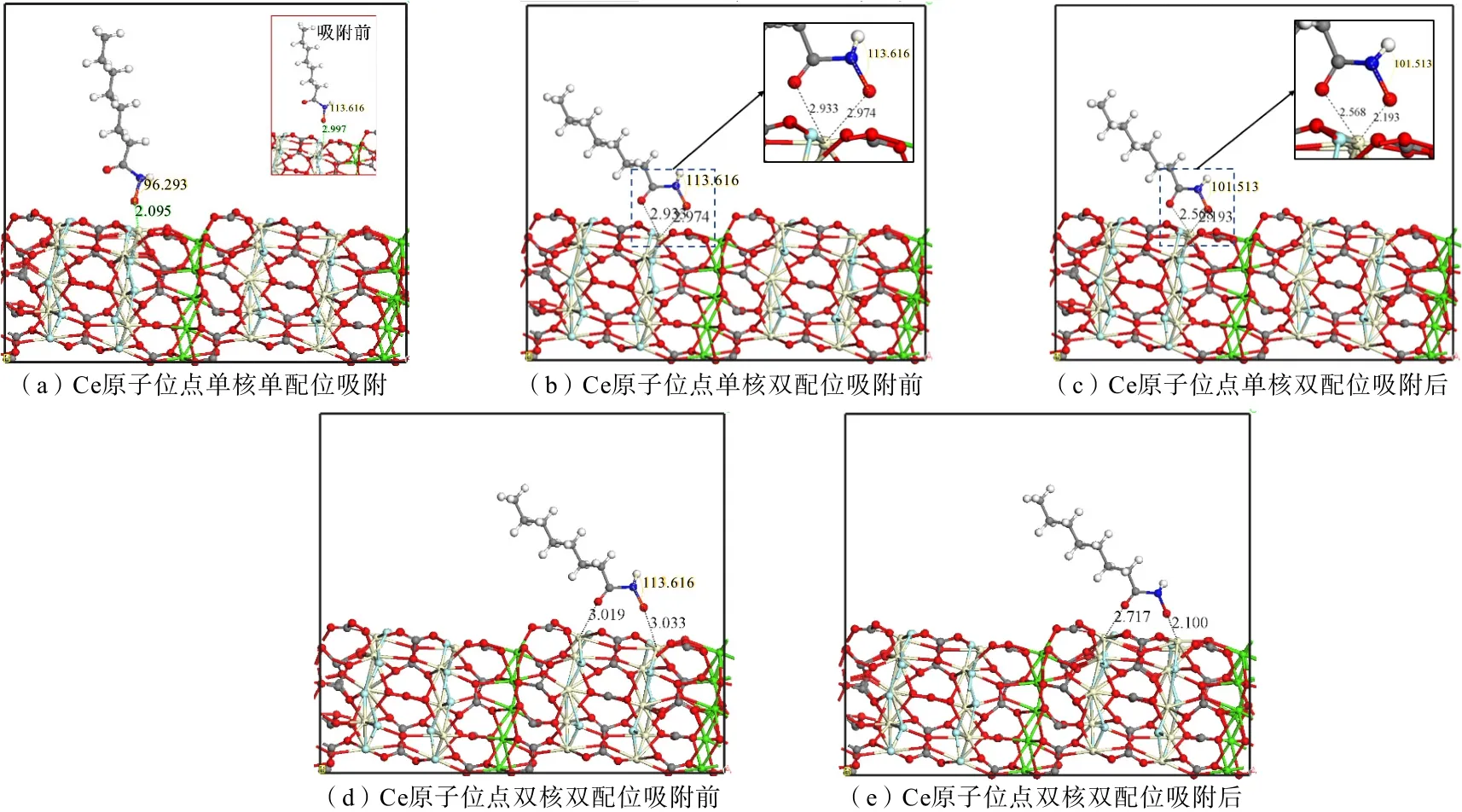
图8 OHA 在氟碳钙铈矿(100)面Ce 位点吸附Fig.8 Configuration for adsorption of OHA at Ce sites on the (100) plane of parisite
由图8 可知,由于OHA 离子组分两个O 具有同等活性,吸附构型分为单核单配位、单核双配位及双核双配位3种,其中单核单配位吸附后,Ce—O 键键长由0.300 nm 缩短为0.210 nm,单核双配位的Ce—O 键键长由0.293、0.297 nm 分别缩短至0.257与0.219 nm,双核双配位的Ce—O 键键长由0.303 nm、0.302 nm 分别缩短至0.210 nm与0.272 nm,表明OHA 离子与Ce 原子成键。结合表3 可知,OHA与Ce 原子3种吸附构型吸附能分别为-600.86、-674.59 及-635.73 kJ/mol,吸附能为负值,表明反应可以自发进行,其中单核双配位吸附构型结合能最低,结构更稳定。
图9所示分别为OHA 在氟碳钙铈矿(100)面单核单配位、单核双配位吸附前后的吸附构型。由图9可知,OHA 离子在Ca 位点吸附形成两种吸附构型,其中OHA与Ca 单核单配位吸附,Ca—O 键键长由0.307 nm 缩短为0.205 nm,单核双配位吸附,Ca—O键键长由0.298 nm与0.301 nm 分别缩短为0.264 nm、0.220 nm,表明OHA 阴离子与氟碳钙铈矿表面Ca 原子吸附成键。由表3 可知,OHA 阴离子在氟碳钙铈矿(100)面Ca 位点吸附能均为负值,表示该吸附过程为自发进行,OHA 阴离子在氟碳钙铈矿(100)面Ca 位点两种吸附构型吸附能分别为-381.64、-507.19 kJ/mol,证明单核双配位形成的五元螯合物构型吸附能更低,构型更稳定。

图9 OHA 在氟碳钙铈矿(100)面Ca 原子位点吸附Fig.9 Configuration for adsorption of OHA at Ca sites on the (100) plane of parisite
综合分析计算结果可知,OHA 阴离子与氟碳钙铈矿表面Ce 位点吸附能力强于Ca 位点,OHA 捕收剂将优先在氟碳钙铈矿表面Ce 位点进行吸附。
3 结 论
(1)DFT 计算表明,氟碳钙铈矿为半导体,(100)面是氟碳钙铈矿的优先解离面。经过优化,氟碳钙铈矿(100)面弛豫较小,表面未发生重构现象。
(2)由于(100)面断键的形成和电荷的再分配,表面Ce、Ca 原子态密度向费米能级偏移,Ce、Ca 原子活性增强,成为荷正电的活性位点,这有利于浮选过程中阴离子药剂在矿物表面的吸附。
(3)OHA 阴离子在氟碳钙铈矿(100)面Ce 原子和Ca 原子位点的吸附构型优化和吸附能计算结果表明,OHA 阴离子能与氟碳钙铈矿表面Ce、Ca 原子吸附成键;OHA 阴离子在氟碳钙铈矿(100)面Ce、Ca位点形成单核双配位吸附构型时吸附能最低,分别为-674.59 kJ/mol与-507.19 kJ/mol,与Ca 位点相比,OHA与Ce 位点形成五元螯合物构型吸附能更低,构型更稳定;OHA 阴离子与氟碳钙铈矿表面Ce 位点吸附能力强于Ca 位点,OHA 捕收剂将优先在氟碳钙铈矿表面Ce 位点进行吸附。
