基于甘松HPLC指纹图谱的模式识别研究及综合主成分评价模型*
2022-11-07杨祎辰王二欢王继强田国庆唐茜琳郭方森王雪健马存德
杨祎辰,王二欢,王继强,田国庆,唐茜琳,张 淼,郭方森,王雪健,马存德
(1.陕西国际商贸学院中药研究院,陕西 咸阳 712046;2.陕西步长制药有限公司,陕西 西安 710075;3.山东步长制药股份有限公司,山东 菏泽 274000)
甘松,别名香松、甘松香,始载于唐·陈藏器的《本草拾遗》[1]。明·李时珍称其“产于川西松洲,味甘”,故而得名[2]。甘松外用祛湿消肿,内服则有理气止痛、开郁醒脾之功效[3]。现代药理临床研究表明,甘松具有抗心律失常、抗心肌缺血、调节血压等作用。Flora of China指出甘松属共2种,分布于喜马拉雅山区,我国产1种,即Nardostachys jatamansi(D.Don)DC.[4],沿用至今。由于甘松适宜的生长环境严苛,人工栽培技术发展滞后,多年以来市场流通供给完全依赖野生采挖。因此,甘松的野生资源破坏严重,濒临灭绝。国内学者对多种濒危药材的质量评价及资源高效利用进行了相关研究,但甘松却少见报道,其研究主要集中在提取分离、临床医学及药理药效学等方向,涉及质量评价相关的研究较少[5]。仅有李莹等[6]、刘国林等[7]及买吾兰江等[8]对甘松的质量评价及特征图谱进行了不同程度的研究,但样本数量少、采集地点单一,且未建立共有模式,亦未能提出有效的质量评价方法,已不能有效反映当前主产区甘松药材的整体质量,亟待深入开展相关研究。此外,基于指纹图谱的综合主成分分析评价,已作为当前中药材质量评价手段之一,广泛应用于丹参[9]、三棱[10]、秦皮[11]等多味大宗中药材,其评价结果具有客观、准确、适用范围广等特点,可弥补甘松药材现代化质量评价手段的空白。基于上述现状,本研究以不同产地收集的甘松药材作为研究对象,通过HPLC法建立甘松药材指纹图谱,并运用主成分分析和系统聚类分析,对所有样品的图谱进行分析并筛选,建立甘松药材指纹图谱;并对其共有模式进行研究,构建甘松药材的综合主成分评价模型,为甘松药材的质量评价提供方法及参考依据,以期规范当前甘松药材的市场流通及产地采收加工现状,保障优质的甘松药材能够应用于中医药临床。
1 仪器与试药
1.1 试药笔者于2021年6—7月,前往四川、甘肃及青海3省,共采集27个批次甘松药材样品。经陕西步长制药有限公司副主任药师马存德鉴定,均为败酱科甘松属甘松Nardostachys jatamansi(D.Don)DC.。样品采集信息表见表1。
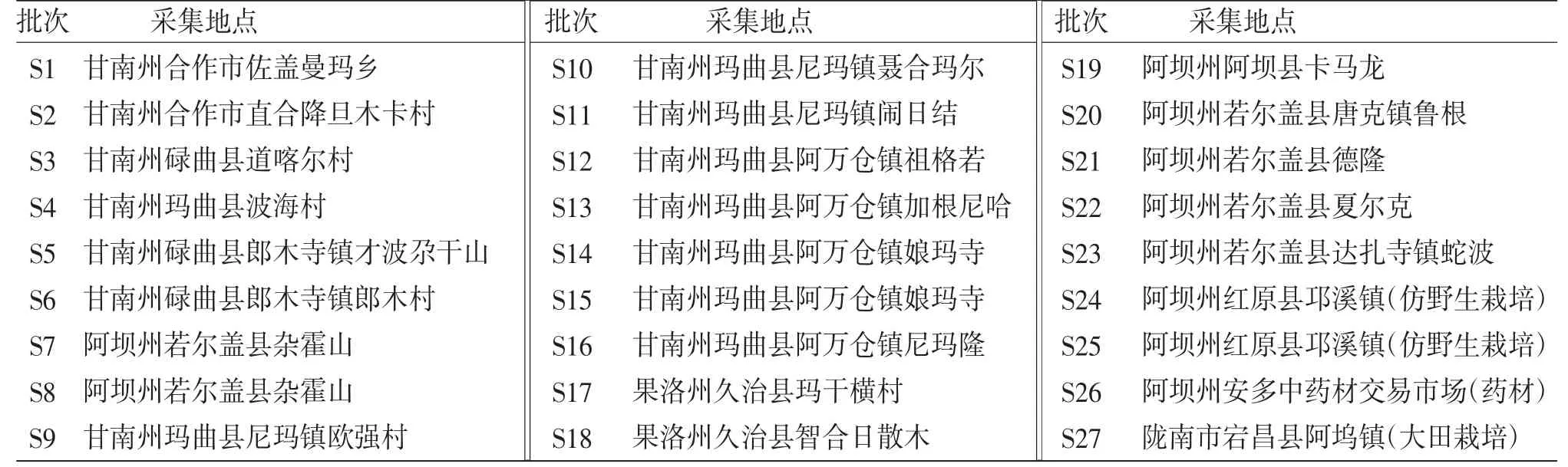
表1 甘松药材样品采集信息
1.2 主要仪器与试剂LC-2030 Plus型液相色谱仪(日本SHIMADZU公司);TU-1810型紫外分光光度计(北京普析通用仪器有限公司);TE124S型分析天平(德国Sartorius公司);QUINTIX224-1CN型电子天平(赛多利斯科学仪器北京有限公司);HC-3018R型高速冷冻离心机(安徽中科中佳科学仪器有限公司);DHG-9050B型电热恒温鼓风干燥箱(上海琅玕实验设备有限公司);KQ-800KDE型高功率数控超声波清洗器(昆山市超声仪器有限公司);TQ-300型高速多功能粉碎机(上海市天祺盛世科技有限公司)。
甘松新酮对照品(批号:AF21021603)、绿原酸对照品(批号:AF21012553)购于成都埃法生物科技有限公司;乙腈、甲醇为色谱纯,购于美国BCR试剂公司;水为娃哈哈纯净水,其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件色谱柱:Diamonsil Plus 5 μm C18-A,250 mm×4.6 mm;流动相:A-(乙腈),B-(0.2%磷酸水溶液);柱温:30℃;流速:1.0 mL/min;进样量:10 μL;检测波长:275 nm。洗脱条件见表2。各主成分与邻峰之间分离度均大于1.5,分离度良好,所测定的各成分理论塔板数均不低于5 000。

表2 洗脱条件
2.2 对照品溶液的制备精密称取甘松新酮对照品适量,加甲醇制成每1 mL中含0.250 0 mg的溶液;精密称取绿原酸对照品适量,加甲醇制成每1 mL中含1.000 1 mg的溶液,即得。
2.3 供试品溶液的制备精密称取甘松药材粉末(过40目筛)0.5 g,置具塞锥形瓶中,准确加入甲醇20 mL,密塞,称定重量,超声处理(功率800 W,频率40 kHz)15 min,放冷,再称质量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.4 方法学考察
2.4.1 线性关系考察 用甲醇稀释对照品溶液,分别为原浓度的1、1/2、1/4、1/8、1/16、1/32倍,摇匀,经0.45 μm滤膜过滤,超声除气,备用。在“2.1”项条件下,以液相色谱峰面积(Y)对两种成分浓度(mg/L,X),绘制标准曲线,分别建立甘松新酮和绿原酸的线性回归方程:Y=31.380X+11.806,R2=0.999 9;Y=37.373X+36.920,R2=0.999 9。表明甘松新酮在7.8~250.0 mg/L浓度范围内、绿原酸在31.2~1 000.0 mg/L浓度范围内呈良好的线性关系。
2.4.2 精密度试验 精密吸取同一供试品溶液(编号:S26),在“2.1”项条件下,连续进样6次。甘松新酮含量分别为1.35%、1.35%、1.32%、1.28%、1.33%、1.43%,平均值为1.34%,RSD为3.74%,绿原酸含量分别为0.15%、0.14%、0.13%、0.14%、0.14%、0.15%,平均值为0.14%,RSD为4.65%。表明仪器精密度良好。
2.4.3 稳定性试验 精密吸取同一供试品溶液(编号:S~26),分别于0、2、4、8、12、24 h在“2.1”项条件下测定。甘松新酮含量分别为1.36%、1.32%、1.32%、1.30%、1.30%、1.30%,平均值为1.32%,RSD为1.64%;绿原酸含量分别为0.15%、0.13%、0.14%、0.14%、0.14%、0.14%、0.13%,平均值为0.14%,RSD为4.79%。表明溶液在24 h内稳定。
2.4.4 重复性试验 精密称取同一样品(编号:S26)6份,按“2.1”项方法制备,在“2.1”项条件下测定。甘松新酮含量分别为1.33%、1.32%、1.33%、1.35%、1.32%、1.30%,平均值为1.32%,RSD为1.09%;绿原酸含量分别为0.13%、0.13%、0.13%、0.14%、0.13%、0.13%,平均值为0.13%,RSD为2.71%。表明方法的重复性良好。
2.4.5 含量测定 分别取27批次不同产地的甘松药材,按“2.3”项方法制备,在“2.1”项条件下测定甘松新酮和绿原酸的含量。
2.4.6 加样回收率试验 取已知质量分数的同一样品(编号:S26)0.5 g,精密称定,平行样9份,分别加入供试品中待测成分含量50%、100%、150%的甘松新酮、绿原酸对照品各3份,混匀;按“2.3”项方法制备,在“2.1”项条件下测定,分别计算甘松新酮和绿原酸的加样回收率。结果显示,甘松新酮、绿原酸的平均回收率分别为97.67%和98.81%,RSD分别为1.46%和2.75%。(见表3)
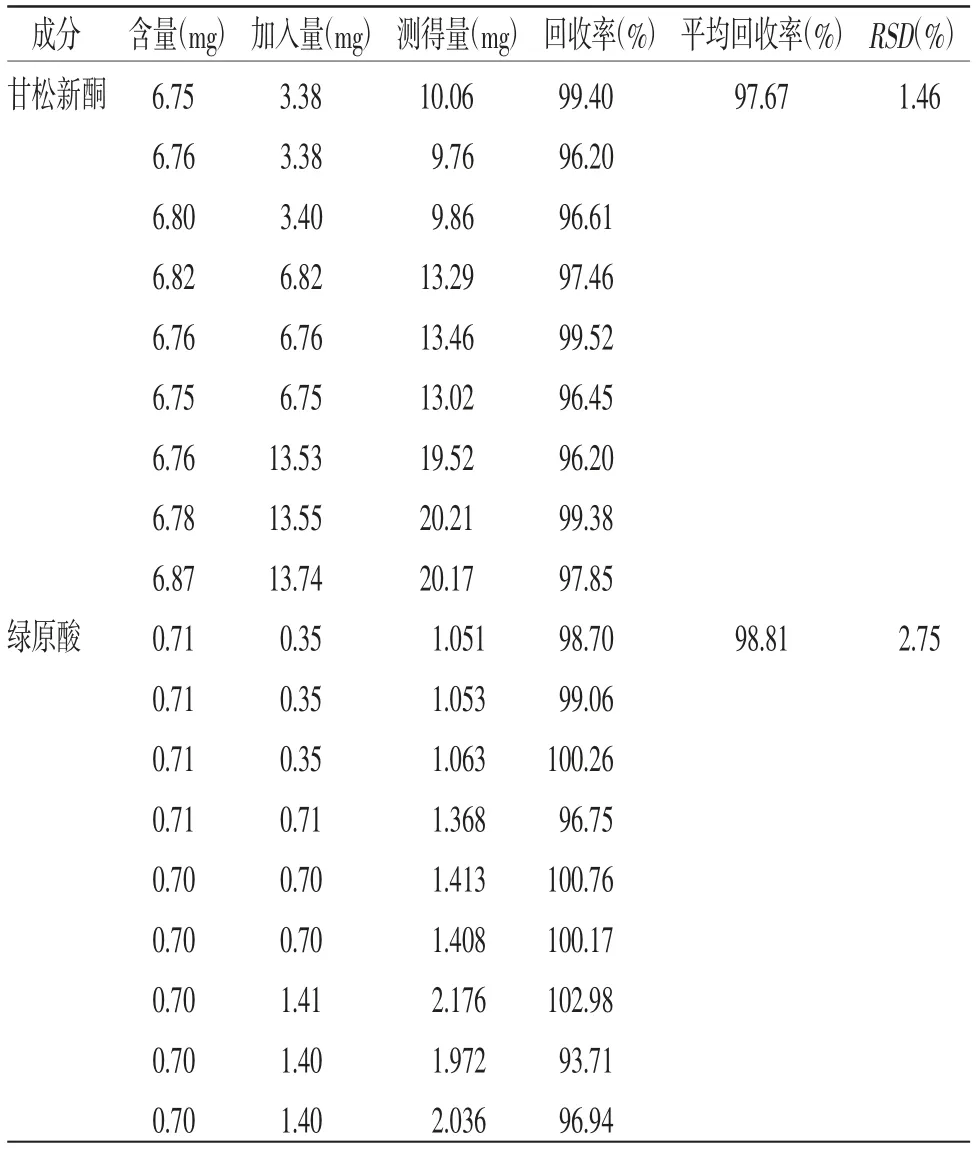
表3 加样回收率试验结果(n=3)
2.5 样品测定绿原酸和甘松新酮为甘松的次生代谢产物,应用于甘松质量评价[12-14]。其中,甘松新酮为现行版《中华人民共和国药典》规定的甘松药材指标性成分。如图1所示,27批次甘松药材的绿原酸含量在0.10%~0.83%范围内,其中S15(甘南州玛曲县阿万仓镇娘玛寺)最高,为0.83%,较最低的S18(果洛州久治县智合日散木)高出730.00%;而甘松新酮含量在0.40%~8.73%范围内,其中S25(阿坝州红原县邛溪镇-仿野生栽培)最高,为8.73%,较最低的S16(甘南州玛曲县阿万仓镇尼玛隆)高出2082.50%。可见,各产地甘松药材的绿原酸和甘松新酮含量差异较大,主要原因考虑如下:(1)甘松属于高原植物,生长条件苛刻,高原地区海拔跨度大、气候各异,不同的生长环境影响植物的次生代谢;(2)甘松为多年生草本植物,本研究收集的样品生长年限未知,导致主要活性成分含量差异较大;(3)甘松样品根茎叶花比例不均一,同种药材的不同部位有效成分含量亦存在一定的差异。现行规定,本品按干燥品计算,含甘松新酮(C15H22O3)不得少于0.10%。本研究收集的27个不同批次甘松药材均高于此一标准。
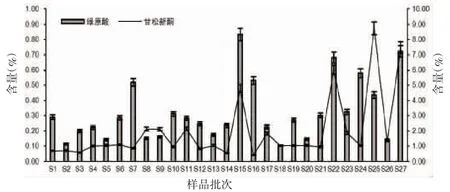
图1 不同产地甘松药材主要活性成分含量
2.6 指纹图谱的建立与化学模式识别分析
2.6.1 甘松药材样品指纹图谱及相似度评价 通过将27批次不同产地的甘松药材样品指纹图谱数据导入《中药色谱指纹图谱相似度评价系统软件》(2012版)软件,并对其进行相似度评价分析。以编号为S26的样品图谱作为参照图谱,筛选出17个主要特征峰,构建甘松药材的指纹图谱,并对2、13号峰进行指认,分别为绿原酸和甘松新酮。后经多点校正,得到27批次甘松样品图谱与对照图谱的相似度计算结果在0.754~0.996之间。其中,S7、S16、S25样品的相似度分别为0.881、0.754、0.896,而其它批次样品的相似度均大于0.900。(见图2)
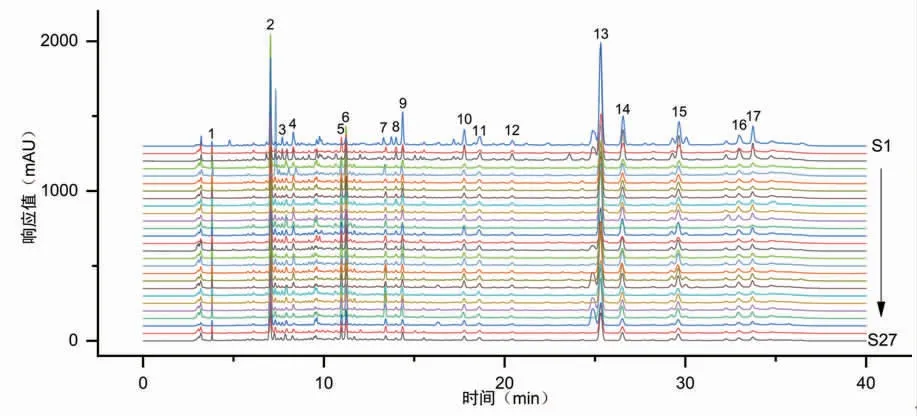
图2 27批次甘松药材指纹图谱
2.6.2 聚类分析 通过将17个共有峰峰面积作为变量,运用SPSS 23.0软件,采用组间联接法,以平方欧式距离27批次甘松药材样品进行系统聚类分析,结果见图3。当组间距离大于10时,27批次样品聚为2类,其中S25(仿野生栽培品)、S27(大田栽培品)为一类,剩余批次为一大类。当组间距离为5~10区间,最多可分为5类。其中,S4、S25、S27分别单独为一类;甘南州的2批(S7、S16)及S24(仿野生栽培品)为一类;剩余批次为一大类,聚类特征较为模糊。当组间距离为0~5区间,该大类中的甘南州的3批(S11、S13、S15)、果洛州的S18及阿坝州的(S20、S23)为一类;S5、S8、S9、S17、S26为一类;甘南州的5批(S1、S6、S10、S12、S14)及阿坝州的2批(S19、S21)为一类;甘南州的2批(S2、S3)为一类;阿坝州的2批(S7、S247)为一类;S22单独为一类;同时,S4、S16、S25、S27分别单独为一类。综上,甘松野生品及栽培品差异明显,而不同产地的甘松亦存在一定的差异。因此,可用聚类分析的方法对药材样品进行分类评价,其结果与相似度评价结果基本保持一致。
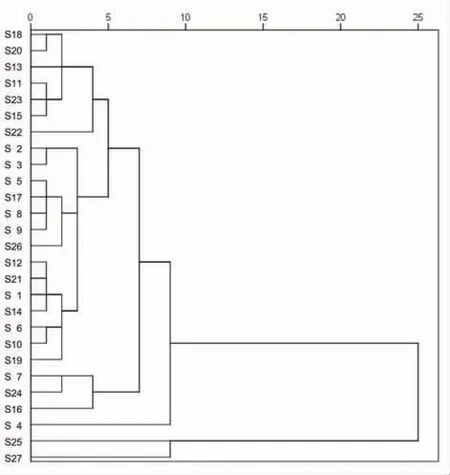
图3 27批次甘松药材的聚类分析
2.6.3 主成分分析(PCA)通过将27批次甘松药材样品的17个共有峰峰面积作为变量,采用SPSS 23.0软件将原始数据矩阵经z-score标准化处理后。通过降维因子分析中的KMO检验和Barlett球形度检验,结果显示:其中KMO取样适切性量数为0.728,适合进行主成分分析;Bartlett球形度检验近似卡方为679.697,自由度为136,P=0.000<0.001,显示该例变量可以为主成分分析提供合理基础。同时,经主成分分析及权重计算后,前3个主成分特征值均大于1,累积贡献率达83.314(见表4);而因子分析碎石图(见图4)亦可看出,在第3个主成分特征值处出现拐点,后逐渐趋于平缓。基于上述分析结果,故提取前3个成分为主成分。基于该条件,以产地作为Group,批次作为Observations,运用OriginPro 2021软件对标准化处理后的原始数据进行主成分分析,并绘制Score Plot图(见图5)。可以看出,除S4、S16、S25、S27外,其余批次药材距离较近且均分别处于两个置信椭圆范围内,与系统聚类分析结果基本一致。
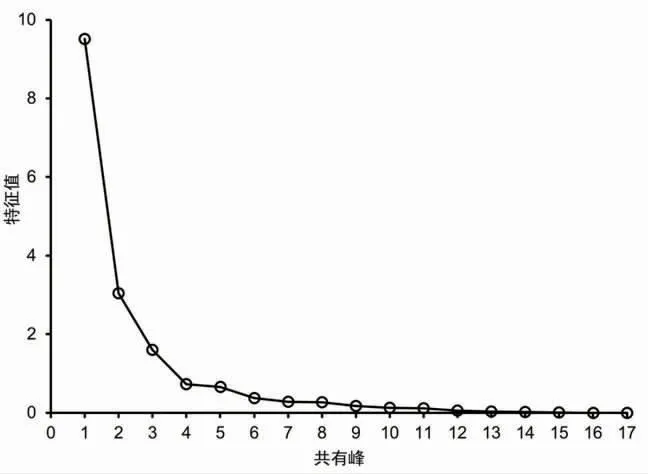
图4 碎石图
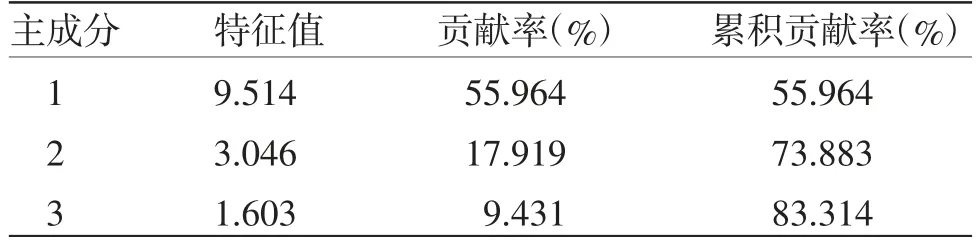
表4 27批甘松共有峰特征值与贡献率
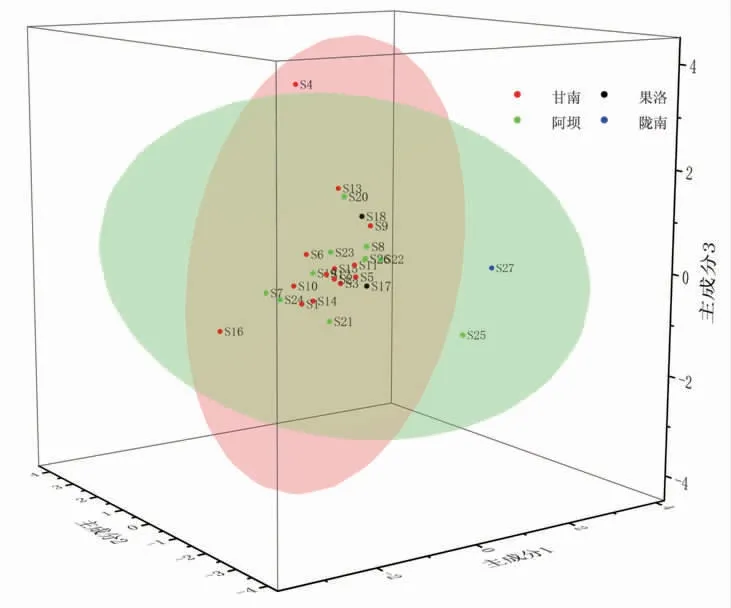
图5 Score Plot图
2.6.4 综合主成分评价模型 由甘松主成分因子负荷矩阵(见表5)可知,主成分1主要解释了F4~F5、F8~F17共13个色谱峰的信息,其中,F5及F8~F17在主成分1中的正相负荷值均大于0.6,即峰面积增加,第1主成分权重亦增大;主成分2主要解释了F1~F3、F6共4个色谱峰的信息,仅有F7在主成分3中的正相负荷值大于0.6。此外,F7~F12、F16共7个色谱峰在全部主成分中负荷值均为正相。通过综合主成分分析,将甘松17个共有峰在各主成分中的因子负荷值转换为对应的线性组合系数(见表5),进而得到主成分1、2、3的评分线性方程Y1、Y2、Y3,再与3个主成分的方差贡献率加权后即得到甘松药材综合质量评价模型F。将z-score标准化处理后的27批次甘松药材样品的17个共有峰峰面积数值依次代入上述方程及评价模型后,结果即为甘松药材综合主成分F值。对F值进行排序,其结果亦是对甘松质量综合评价的结果。(见表6)
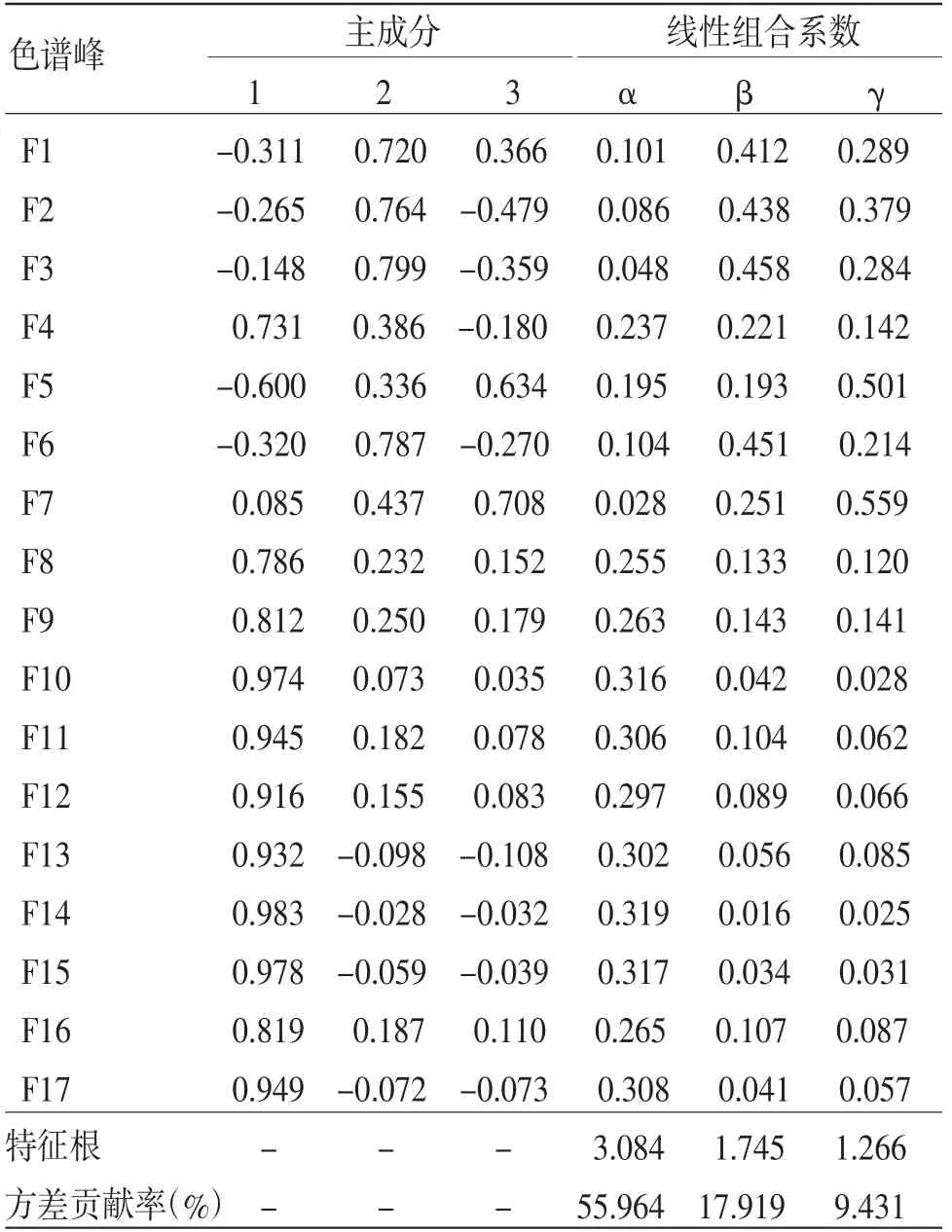
表5 主成分分析因子负荷矩阵及线性组合系数
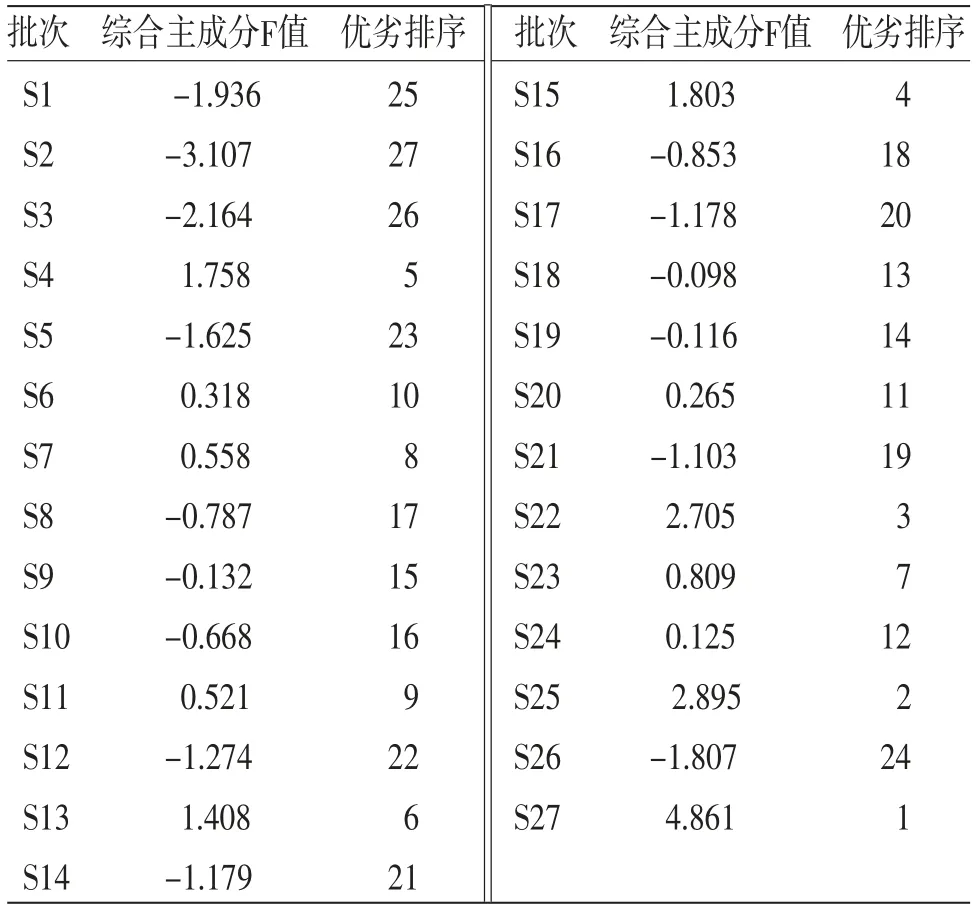
表6 综合主成分评价结果
计算公式如下:

综上可知,主成分因子得分值计算公式为Yi=∑Xj·λj,而综合主成分评价模型F=∑Yi;式中,Xj为标准化后的峰面积值,λj为线性组合系数。
2.6.5 甘松药材标准指纹图谱的建立 基于相似度评价,参考主成分分析及系统聚类分析结果,将S4、S16、S25、S27批次剔除,取剩余23批次甘松样品建立甘松药材的标准指纹图谱。将23批次甘松样品指纹图谱数据重新导入《中药色谱指纹图谱相似度评价系统》(2012版)软件,对其进行相似度评价(见表7);并继续设定S26为参照图谱,经多点校正、匹配,R为生成的对照指纹图谱(见图6)。结果显示,剩余23批次甘松样品与对照图谱比较的相似度均在0.915以上,这可为甘松药材的质量评价提供科学依据。
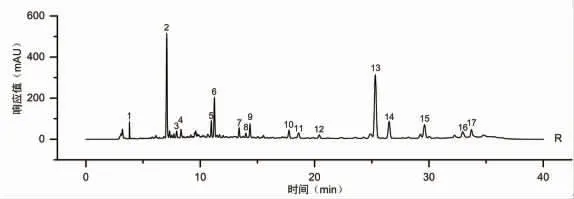
图6 甘松药材标准指纹图谱
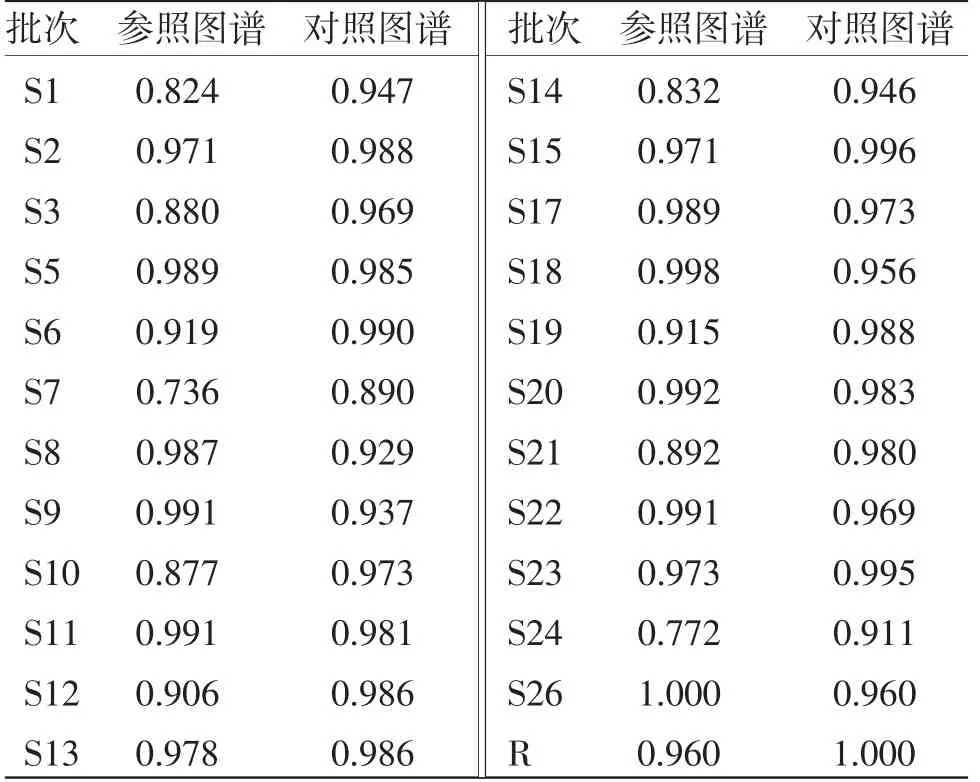
表7 23批次甘松样品的相似度评价
3 讨 论
3.1 供试品的制备方法参照2020年版《中华人民共和国药典》甘松新酮“含量测定”项下供试品溶液的制备方法[3],在超声处理(功率50 W,频率45 kHz)15 min条件下,开展重复性试验时,RSD较大。考虑为超声提取不充分,故采用超声处理(功率800 W,频率40 KHz)15 min,结果较好。此外,2020年版《中华人民共和国药典》规定甘松的入药部位为根及根茎,而本研究所采集的甘松样品均为全草。原因在于甘松资源均依赖于野生,且资源量匮乏,实际流通品均为全草。因此,结合市场及产地流通调研情况及文献报道,本研究中各批次样品检测部位均为甘松全草。
3.2 色谱条件的选择为了保证建立的甘松指纹图谱能够体现更多的化学成分,且筛选的2种有效成分具有较好的分离度,使得该方法适宜于定量分析。故分别对绿原酸和甘松新酮对照品溶液在紫外190~400 nm波长范围内进行全波长扫描,发现其最大吸收波长差异较大。综合考虑,选择在275 nm波长条件下对2种成分进行测定,结果较好。此外,配置2种成分的混合对照品溶液考察洗脱体系,结果发现以乙腈-0.2%磷酸水溶液作为流动相,对混合对照品的分离度较好,基线稳定,色谱峰理论塔板数高,同时也适用于供试品溶液。
买吾兰江等[8]建立的甘松药材HPLC指纹图谱方法需60 min,且30 min后已无共有特征峰出现,仅标注4个共有峰;而李莹等[5]以10批药材,建立的甘松HPLC指纹图谱方法则需65 min,但色谱峰分离度一般。本研究构建的HPLC指纹图谱,分析时间短(40 min),减少了溶剂用量,降低了分析及人工成本,同时各色谱峰分离效果良好。
3.3 分析方法评价23批次药材与对照指纹图谱相似度均在0.915以上,表明不同产地的甘松药材质量较稳定。主成分分析结果与聚类分析结果基本一致,大多数甘松野生品聚为一类,而甘松栽培品聚为一类,但其中亦有个别产地的甘松野生品与栽培品未能有效聚类。考虑由于部分产地的栽培甘松采用仿野生栽培模式,因此质量与野生品较为接近。此外,由于甘松属多年生草本,而此生代谢产物的积累往往与植物的生长年限相关,野生甘松生长年限等信息的不明确,亦会影响聚类分析的结果。因此,尚需开展不同采收期栽培甘松或不同生长年限的野生甘松质量评价研究。综上,建立的分析方法基本可行,亦可为甘松的来源及产地识别提供支持。
3.4 综合主成分评价模型指纹图谱是中药材质量评价、控制的主要手段之一,其可通过相似度评价体现不同批次药材的差异;但当样本量过大且相似度较为接近时,单一的指纹图谱便不能有效、准确地判断药材质量的优劣,往往需要结合统计学评价[15-17]。而综合主成分评价法是在主成分分析的基础上,在不损失或尽量少损失原样本内在指标信息的前提下,将数个具有一定相关性的指标通过加权计算,转换成一个综合指标F值,以此来进行综合评价、考量,并据此对不同样本进行优劣排序。本研究建立的综合主成分评价模型结果表明,S27、S25的F值最高,分别为陇南大田栽培品及阿坝仿野生栽培品,而系统聚类分析中当组间距离大于10时,S27、S25亦聚为一类,其它批次为一大类,二者结果保持一致;S2、S3的F值最低,均为甘南合作的野生品,而系统聚类分析中当组间距离小于3时,S2、S3聚为一类,故综合主成分评价模型得到了系统聚类分析的验证。因此,基于甘松药材HPLC指纹图谱构建的综合主成分评价模型较好,具有一定参考价值。
4 小 结
本研究通过HPLC法建立了不同产地甘松药材的指纹图谱,通过主成分分析及系统聚类分析两种统计学方法对色谱数据进行模式识别初探。基于分析结果,剔除了个别样品,确定入选的样品批次,最终得到了较为标准的甘松药材对照指纹图谱,为甘松药材的质量评价研究奠定了基础。此外,首次建立了基于指纹图谱的甘松药材综合主成分评价模型。其评价结果与系统聚类基本保持一致,并能够较好地体现相似度评价,其F值的优劣排序具有一定的客观性,可作为未来甘松药材质量评价的手段。同时,研究结果亦表明不同产地的野生甘松药材、野生与栽培甘松药材的质量均具有明显的差异。因此,基于本研究取得的成果,亟待进一步开展甘松野生与栽培品主要活性成分及不同产地甘松的主要活性成分含量研究,为甘松的产地与品质的相关性及甘松药材的道地性提供理论依据和数据支撑[18-21]。但本研究亦有不足之处,由于全部27批次甘松样品生长年限等信息不明确,导致主要活性成分含量测定差异较大,未能得到与产地的相互关联性分析,具有一定的局限性,还需进一步优化样品采集方式,深入开展相关研究。
