烧结烟气中含钾化合物对钒钨钛催化剂脱硝/二噁英性能的影响
2022-11-06钱立新张洪亮魏进超杨本涛龙红明
丁 龙,杨 涛,钱立新,张洪亮,魏进超,杨本涛,龙红明
1) 安徽工业大学冶金工程学院,马鞍山 243032 2) 中冶长天国际工程有限责任公司,长沙 410205 3) 冶金工程与资源综合利用安徽省重点实验室(安徽工业大学),马鞍山 243002
铁矿烧结是钢铁冶金长流程生产中重要的工艺之一,其生产过程会产生包括二氧化硫(SO2)、氮氧化物(NOx)和二噁英等在内的多种污染物[1-3].其中,NOx会造成环境中光化学污染和酸雨等问题,也会刺激生物体呼吸系统,造成胸闷、咳嗽等症状;二噁英是一类毒性强、易致癌和致畸的持久性环境激素类有机物[4-5].考虑到NOx和二噁英对人类健康和生态环境的危害,控制这两类污染物的产生与排放一直是企业和研究者非常关注的问题.随着国家环境治理力度不断加强,特别是2019年中国钢铁工业实施烟气污染物超低排放[3],要求铁矿烧结烟气中NOx排放限值由300 mg·m-3降为50 mg·m-3以下,二噁英的排放限值也建议控制在(毒性当量)0.1 ng·m-3以下.因此,烧结烟气多污染物中温协同催化净化技术得到快速发展,并逐渐成为烧结烟气中脱硝/二噁英应用最广泛的技术之一[6].
烧结烟气多污染物中温协同催化净化技术的核心是催化剂,目前工业应用最为成熟、广泛的是V2O5-WO3(MoO3)/TiO2(VWTi)催化剂[7].VWTi催化剂一般由活性成分、活性助剂、载体及其他提供粘结、填充功能的辅助成分组成.活性成分主要是V2O5,WO3(或MoO3)是活性助剂,载体主要是TiO2[8-9].国内较早采用VWTi 催化剂进行脱硝的是上海宝山钢铁股份有限公司[10],他们在烟气脱硫工艺后建设了一套中温脱硝装置,通过加热炉先将烟气温度加热到280 ℃左右,再进行NOx减排.该脱硝系统投产运行后,NOx排放质量浓度最低减排到34 mg·m-3,满足国家超低排放要求.
“十四五”期间,国内钢铁企业的烧结烟气逐步采用VWTi 催化剂进行污染物减排.烧结烟气中含有的粉尘、有害组分会与催化剂接触,导致催化剂活性降低,最终失活形成废弃物.目前,大量研究表明[11-13],工业烟气中应用的VWTi 催化剂失活方式主要包括物理失活和化学失活.当灰分和有害成分仅覆盖在催化剂的活性位点上,或者造成催化剂孔道堵塞的失活通常被称为物理失活,物理失活的催化剂通常可以通过去除堵塞物进行再生.当有害成分与催化剂表面活性中心发生化学反应,并造成其活性位点化学环境改变的失活通常被称为化学失活,这也是造成催化剂变成废弃物的主要原因之一.目前,燃煤电厂催化剂失活研究表明,烟气中致催化剂失活的有害成分包括碱金属[14]、碱土金属[15]、类金属(通常指砷)[16]以及重金属[17]等.其中,碱金属在烟气中含量较高,被认为是导致催化剂失活的主因.对于碱金属造成催化剂的失活机理,前人研究表明主要是催化剂表面的酸性位点与金属离子发生了反应,导致活性中心改变,影响了还原剂NH3在催化剂表面的吸附和转化,从而导致催化剂失活[18].
烧结烟气同样也是一种多污染物与有害元素并存的工业烟气,研究表明[19-20],烧结烟气中碱金属元素质量浓度在24~60 mg·m-3,碱金属元素在除尘灰中质量分数更是达到5%~40%,且主要以钾元素为主,占比达到80%以上.虽然研究者已经开展了大量关于不同形态钾盐对VWTi 催化剂失活机理的研究,但以往的研究多是基于燃煤烟气特点,研究不同形态钾盐负载对VWTi 催化剂脱硝性能的影响.烧结烟气是一种多污染物并存的烟气,VWTi 催化剂在烧结烟气中应用,既可以催化NOx还原,也可以催化二噁英类有机物氧化[21].然而,目前很少有报道基于烧结烟气特点,研究不同形态钾盐负载对VWTi 催化剂同时脱硝脱二噁英性能的影响,催化剂的失活机理也有待进一步明确.
本文模拟烧结烟气特点,选用商用VWTi 催化剂为原料,在实验室模拟工业现场催化剂加速失活条件,研究了VWTi 催化剂表面负载KCl、K2O和K2SO4对其同时脱硝脱二噁英活性的影响,并通过比表面积检测(BET)、X 射线衍射(XRD)、氢气程序升温还原(H2-TPR)、氨气程序升温脱附(NH3-TPD)、X 射线光电子能谱(XPS)、红外光谱分析(FT-IR)等手段研究了以上三种钾盐对催化剂结构、表面酸性、表面活性物质化学环境及氧化还原性能的影响.针对失活的催化剂,研究了水洗和酸洗两种再生手段对其活性修复效果.最后提出了不同形态钾盐致VWTi 催化剂失活的机理.
1 实验材料及实验方法
1.1 实验材料
本研究的实验材料包括催化剂和中毒物质,选用的催化剂为市场商用蜂窝状VWTi 催化剂,该催化剂主要用于钢铁企业烧结厂烟气脱硝脱二噁英,催化剂的化学成分如表1 所示,TiO2为催化剂的载体,V2O5是催化剂的活性组份,WO3是催化剂的助剂,其他组份为催化剂成型过程添加的粘结剂、造孔剂等.中毒物质选用K2SO4、KNO3和KCl 化学纯试剂,其中,KNO3作为K2O 中毒的前驱物.

表1 SCR 催化剂化学成分(质量分数)Table 1 Chemical composition of the SCR catalyst (mass fraction) %
1.2 中毒催化剂制备及其再生方法
在实验室模拟工业现场条件,进行了商用VWTi催化剂加速失活实验.通过浸渍蒸干法将一定质量分数的钾盐负载到新鲜催化剂表面得到预中毒催化剂,之后在空气条件下煅烧得到不同类型K中毒催化剂样品.
KCl 中毒催化剂样品的制备实验步骤如下:首先,将新鲜VWTi 催化剂进行研磨,得到颗粒粒径小于0.250 mm 的催化剂粉末,置于105 ℃烘箱中烘干备用,记为Fresh.然后,按照KCl 与新鲜催化剂质量比为1∶50,称取质量分数为2.0%的KCl 溶于100 mL 去离子水中,并向溶液中加入干燥的新鲜催化剂粉末.混合物在70 ℃条件下水浴搅拌4 h,并于105 ℃的烘箱中干燥12 h.将中毒催化剂研磨成粒径小于0.250 mm 的粉末,置入马弗炉中,在500 ℃条件下焙烧5 h,得到KCl 中毒的催化剂样品,记为KC.以同样的方法制备K2O、K2SO4中毒催化剂,记为KO、KS(KNO3的称取量需要换算成对应质量分数的K2O).将中毒的催化剂粉末样品压成小饼,制取粒径为0.250~0.425 mm 颗粒进行活性检测.最后,将中毒催化剂样品进行性能表征和再生实验.新鲜催化剂与中毒催化剂按照相同焙烧制度进行焙烧处理.
本文对中毒催化剂采用了两种常见的活性再生方式处理,即水洗和酸洗.
(1)水洗再生:将一定量的中毒催化剂样品,按照固液比1∶20 放入去离子水中洗涤,洗涤时间30 min,搅拌速度500 r·min-1,温度25 ℃,洗涤结束后对再生催化剂样品进行过滤,在105 ℃干燥箱中进行干燥12 h,并在马弗炉中空气条件下500 ℃煅烧3 h.
(2)酸洗再生:配制质量分数为1%的H2SO4溶液,将一定量的中毒催化剂样品,按照固液比1∶20 放入弱酸溶液中进行洗涤,其他实验过程与水洗再生方法相同.
1.3 催化剂活性评价方法
采用固定床反应器进行配气模拟烧结烟气特性,研究新鲜和中毒催化剂同时脱除NO 和二噁英的活性.由于二噁英结构复杂,毒性强,分析过程复杂,研究者在实验阶段通常采用化学结构与二噁英相近的氯苯(CB)为二噁英模拟物,用于模拟二噁英进行催化活性检测[22-23],因此,本研究同样选用CB 作为二噁英替代物.催化反应装置示意图如图1 所示.入口混合气体的体积分数分别为:NO,0.03%;NH3,0.03%;CO,0.50%;O2,16%;CB,0.01%.气体总流量为160 mL·min-1,空速比(GHSV)为60000 h-1,N2作为平衡气体,其中,CB 采用N2鼓泡方式通入混合气体中.实验前调整设定气体,并将0.2 g 催化剂样品装填到石英管中,检测温度在150~450 ℃之间,间隔50 ℃,当温度升高至设定值时,系统温度稳定20 min 后,收集出口气体15 min,并采用便携式烟气分析仪(RBR,德国ECOM J2KN)检测NO 体积分数,采用气相色谱仪GC-9860 在线分析CB 体积分数.

图1 催化剂活性测试反应装置Fig.1 Schematic of the laboratory activity test of catalysts
通过NO 转化率(ηNO)计算不同催化剂的脱硝活性,计算公式为:

式中:V[NO]in为进气处烟气中NO 的体积分数;V[NO]out为出口处烟气中NO 的体积分数.
通过CB 转化率(ηCB)计算不同催化剂的CB 催化降解活性,计算公式为:

式中:V[CB]in和V[CB]out分别是反应器进出口的CB体积分数.
1.4 催化剂物化性能评价方法
对中毒催化剂和新鲜催化剂进行物化性能评价.采用X 射线荧光光谱分析仪(XRF,美国,ARLAdvant’X IntellipowerTM3600)测定催化剂样品各元素含量;X 射线衍射分析仪(XRD,德国,D8ADVANCE)测定样品晶体结构;比表面积与孔隙度吸附仪(BET,美国,ASAP 2460)测定催化剂样品的比表面积及孔结构;X 射线光电子能谱仪(XPS,日本,PHI Quantera Ⅱ)测定催化剂表面各元素化学结构;傅立叶红外光谱仪(FTIR,美国,Nicolet6700)分析催化剂样品表面化学键;氨气程序升温脱附(NH3-TPD)和氢气程序升温还原(H2-TPR)实验,分别在N-3000 双通道色谱工作站设备上进行,测定催化剂表面活性位点和催化剂表面活性物质氧化还原性.通过以上催化剂性能表征手段,可以获得催化剂中毒前后结晶形式、催化剂形貌、孔结构参数、表面元素价态、氧化还原性能、表面官能团分布等变化,进而揭示催化剂的失活机理.
2 结果与讨论
2.1 不同形态钾盐对催化剂性能的影响
2.1.1 对催化剂脱除NO 和CB 活性的影响
新鲜及不同形态钾盐中毒催化剂的NO 和CB 降解活性如图2 所示.从图中2(a)可以看出,在空速比60000 h-1下,新鲜催化剂展现出良好的脱硝活性,在250~400 ℃温度范围内的脱硝效率接近100%.引入不同形态钾盐后,催化剂活性出现了不同程度的降低,当KCl 加入到催化剂中,最高脱硝效率出现在350 ℃,仅为50%.当K 盐以K2O的形式引入时,最高脱硝活性为72%,该值在300 ℃时获得.相比以上两种K 盐存在形式,K2SO4对催化剂的脱硝活性影响最小,其最高活性为84%,在300 ℃时获得.从图2(b)中可以看出,新鲜催化剂和中毒催化剂的CB 降解活性均随着温度升高而升高,均在450 ℃获得最高活性.在新鲜催化剂表面负载三种钾盐后,催化剂CB 降解活性出现了显著降低,在450 ℃时,Fresh、KS、KO 和KC 样品的CB 降解效率分别为96.3%、25.1%、22.2%和8.4%,可以看出,中毒催化剂样品几乎失去了对CB的催化降解活性.综合以上结果,不同形态钾盐引入对催化剂的脱硝和CB 降解活性损失顺序遵循相同的规律,即KCl> K2O> K2SO4.

图2 不同形态钾盐中毒对催化剂活性的影响.(a) 脱硝活性;(b)CB 降解活性Fig.2 Catalytic activity of catalysts poisoned by different K+ species:(a) NO conversion;(b) CB conversion
2.1.2 对催化剂结构特征的影响
通过对新鲜和中毒催化剂的BET 比表面积和孔结构参数分析,可以考察不同形态钾盐对催化剂的物理堵孔效应.由表2 可知,新鲜催化剂的比表面积和总孔体积数值在三种钾盐引入后均有不同程度的降低,平均孔径变大,这主要是由于钾盐堵塞了催化剂的孔结构.相比之下,负载K2SO4对催化剂的比表面积影响最大,表明堵孔效应最显著.然而,结合催化剂的脱硝和CB 降解活性,虽然硫酸盐对催化剂的孔结构影响最大,但其对催化剂的活性影响却是最小的.因此,负载不同形态钾盐对催化剂比表面积带来的变化并不是引起其活性降低的主要因素,即物理失活作用应不是钾盐致催化剂活性显著下降的主因,研究化学失活应比物理堵孔更为重要.

表2 新鲜和中毒催化剂样品的物理吸附参数分析Table 2 Physical adsorption parameters of fresh and poisoned catalysts
新鲜和不同形态钾盐中毒催化剂的XRD 图谱如图3 所示.由图可见,新鲜催化剂和负载不同形态钾盐获得的XRD 图谱上仅出现了锐钛矿型TiO2和成型剂SiO2、Al2O3的衍射峰,没有发现V2O5、WO3、K2SO4、K2O 以及KCl 的衍射峰,说明引入不同形态钾盐并没有造成催化剂活性物种分散度改变,活性物种没有形成相应的结晶态,且不同形态钾盐在催化剂表面可能以高度分散形式存在,没有形成相应的结晶态.
2.1.3 对催化剂氧化还原性能的影响
为了研究引入不同形态钾盐对VWTi 催化剂氧化还原性能的影响,通过H2-TPR 实验对新鲜和中毒催化剂进行了表征,结果如图4 所示.可以看出,新鲜催化剂在200~750 ℃范围内出现了一个还原峰,峰中心位于472 ℃处,该峰可以归为V5+到V3+的转化[18].一般认为,V5+的还原峰温度越低,越有利于催化反应的循环过程.在新鲜催化剂上负载不同形态钾盐后,钒物种的H2还原峰均向高温方向偏移,且偏移量顺序为KC> KO> KS,这与催化剂的脱硝和CB 降解活性降低幅度顺序一致.还原峰向高温方向移动意味着催化剂中钒物种更难被还原,即催化反应中,催化剂氧化还原能力下降,进而导致其脱硝和CB 降解活性减弱.这主要是由于钾物种与催化剂表面的活性物质反应,进入了其晶格中,形成了强化学键,导致催化剂中晶格氧含量降低或者抑制了晶格氧在反应中的释放,从而造成钒物种难以被还原.

图4 不同形态钾盐负载后催化剂的H2-TPR 图谱Fig.4 H2-TPR profiles of catalysts poisoned by different K+ species
2.1.4 对催化剂表面酸性的影响
长时间以来,研究者普遍认为VWTi 催化剂的表面酸性是其脱硝和催化降解二噁英的重要指标[24],为了解释新鲜和中毒催化剂表面酸性变化对催化还原NO 和催化氧化CB 活性的影响,对新鲜和中毒样品进行了NH3-TPD 分析,结果如图5所示.新鲜催化剂在100~550 ℃的温度范围内出现了两个NH3脱附峰,分别对应于弱(低温)和强(高温)结合NH3的峰.弱结合NH3的峰归属于Brønsted 酸性位点,强结合NH3的峰归属于Lewis 酸性位点[15,25].酸性位点主要作用是为参与催化反应的物质提供反应位置.催化过程中所发生的氧化还原,均在酸性位点上完成,酸性位点的数量会影响催化剂活性.
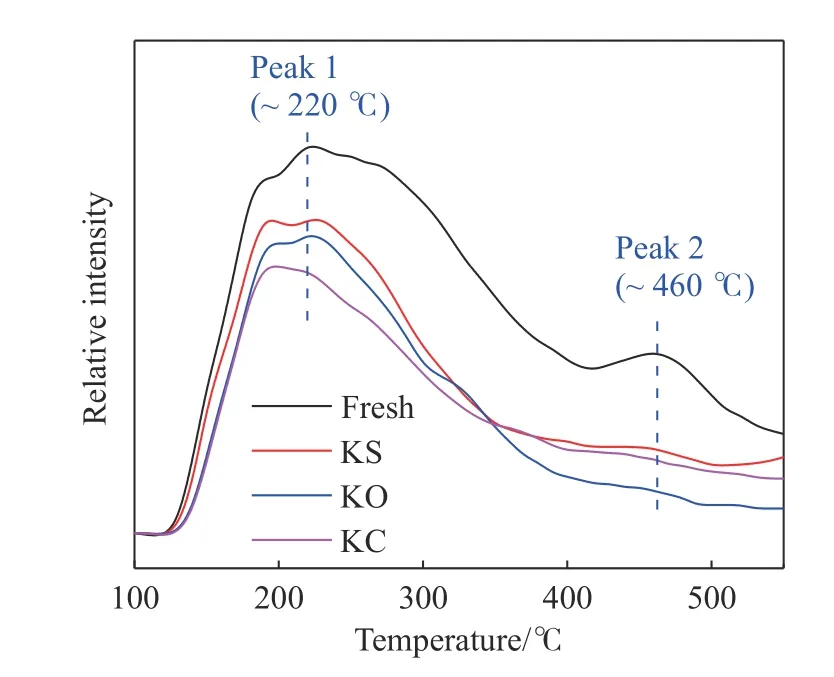
图5 不同形态钾盐负载后催化剂的NH3-TPD 图谱Fig.5 NH3-TPD profiles of catalysts poisoned by different K+ species
从图5 中可以看出,负载三种钾盐后,220 ℃附近的Brønsted 酸性位点的NH3脱附峰强度明显降低,降低幅度顺序为KC>KO>KS,这与催化剂的活性降低顺序相吻合.相比之下,三种钾盐对460 ℃附近的Lewis 酸性位点产生了更大的影响,NH3脱附峰几乎消失,其中K2O 的影响最大,其次是KCl,影响程度遵循KO>KC>KS,这与Kong 等[18]研究结论相似.主要是因为引入KCl 后,Cl-可以结合在催化剂表面,增强催化剂对NH3的吸附能力,但Cl-吸附NH3后并不能将其活化,这部分NH3不参与选择性催化还原(SCR)反应,也就是Cl-形成的酸性位点为“伪酸性位点”.对于CB 降解反应,催化剂表面结合Cl-后,会降低催化剂表面酸性位点对CB 分子中C—Cl 键的亲核取代吸附作用,也就是催化剂发生了Cl 中毒.虽然K2SO4中的K+会与催化剂表面的酸性位点结合,造成NH3吸附量减少,但SO42-具有弱酸性位性能[26-27],它可为脱硝和CB氧化反应提供新的酸性位点,这可能是K2SO4中毒的催化剂样品仍然表现出较高的活性的原因.
2.1.5 对催化剂表面活性中心化学环境的影响
根据H2-TPR 结果,催化剂表面引入不同形态钾盐后,钒物种的氧化还原能力降低,而氧化还原能力与催化剂表面活性物种的化学形态和表面氧元素含量息息相关.对新鲜和中毒催化剂样品进行了XPS 分析,结果如图6 所示.图6(a)显示了Ti 2p的XPS 图谱,根据文献报道,Ti 2p 结合能在464.6 eV(Ti 2p1/2)和458.8 eV(Ti 2p3/2)处均对应锐钛矿型TiO2的特征峰[28].从图中可以发现,KS、KO 和KC样品的结合能与新鲜催化剂相比没有发生明显变化,说明不同形态钾盐的引入没有改变TiO2载体的化学环境,这与XRD 的结果相吻合.图6(b)为W 4f 的XPS 图谱.W 4f 结合能在不同形态钾盐引入前后也未发生明显变化,表明K 并未与催化剂表面W 结合,没有对W 的化学环境产生影响.
在Topsøn[29]提出的SCR 脱硝反应机制中,V5+=O 在循环反应中起到关键作用,且V5+=O 是CB 亲核取代实现C—Cl 键断裂反应的重要活性位点.因此,V5+比例将直接影响催化剂的脱硝和CB 降解性能,采用V5+/V4+摩尔比来反应催化剂的反应性能.图6(c)显示了V 2p 的XPS 图谱,表3 汇总了不同形态钾盐负载后催化剂样品V 元素XPS 结果(表中Ebv表示结合能值,ω表示质量分数).对V 谱图进行分峰和拟合,结合能在517.4、516.2、515.3 eV 分别对应V5+、V4+、V3+的结合能特征峰[30].综合来看,负载不同形态钾盐后,催化剂表面V5+/V4+摩尔比有所降低,由新鲜催化剂的1.97 降低到KS 的1.30、KO 的1.26 和KC 的1.15,降低幅度与催化剂活性降低程度保持一致,V5+/V4+摩尔比的降低会直接减弱催化剂的氧化还原性能,从而降低催化剂的活性.结合NH3-TPD 结果,这可能是钾离子进入催化剂表面的晶格,与V5+=O 键相互作用形成了V4+—O—K,造成V5+比例降低.

表3 不同形态钾盐负载后催化剂样品V 元素XPS 结果Table 3 XPS results of V of fresh and poisoned catalyst samples
图6(d)是O 1s 的XPS 图谱,根据O 1s 的分峰结果,氧原子的能谱峰可以分为两部分,结合能在532.2 eV 附近的为表面吸附氧(以Oα表示),在529.9 eV 附近的为晶格氧(以Oβ表示)[31].通常,表面吸附氧在VWTi 催化剂的氧化还原反应中具备比晶格氧更好的移动能力,在脱硝过程中,表面吸附氧以O2-、O-、O2-形式存在,将吸附NH3氧化成—NH4+,参与后续脱硝反应.在CB 氧化过程中,表面吸附氧持续攻击CB 中C—Cl 键,使其断裂,生成苯酚类物质,以及Cl-.苯酚类物质中的π 键继续被表面吸附氧氧化开环,最终氧化成CO2和H2O 等物质,完成降解,因此表面吸附氧参与氧化反应时能发挥更重要的作用.研究者们通常用Oα/(Oα+Oβ)质量分数比值来衡量催化剂中Oα的含量,并考察其与催化剂活性的相互关系.由图5(d)的分峰面积结果计算,新鲜催化剂的表面化学吸附氧质量分数为41.5%,而不同形态钾盐中毒后,对应催化剂表面化学吸附氧百分含量均有不同程度的降低,比例遵循KS> KO> KC,与催化剂活性变化结果一致,这主要是由于K+与催化剂表面化学吸附氧发生反应所致.

图6 不同形态钾盐负载后催化剂的XPS 图谱.(a) Ti 2p;(b) W 4f;(c) V 2p;(d) O 1sFig.6 XPS profiles of catalysts poisoned by different K+ species: (a) Ti 2p;(b) W 4f;(c) V 2p;(d) O 1s
2.1.6 对催化剂表面活性官能团的影响
在VWTi 催化剂脱硝过程中,NH3首先被V—OH 吸附反应形成-NH4+,再被V5+=O 转化形成NH3—V5+=O,然后再吸附NO 形成中间过渡物NO-NH2-V4+-OH,最后中间过渡物质分解形成N2和H2O,同时在氧气存在的条件下,V4+—OH 被缓慢氧化回V5+=O[32].而在CB 降解过程,CB 分子首先吸附在催化剂表面,并在V=O 活性位点作用下发生亲核取代反应,使得C—Cl 键断裂,形成酚类物质.酚类物质通过亲电取代作用,与催化剂中氧(O2-、O-、O2-)发生氧化反应,形成苯醌类物质,该物质进一步氧化开环,并形成马来酸盐、乙酸盐和氯乙酰基物质,最终CB 被氧化形成CO、CO2、HCl 和H2O 等从催化剂表面脱除[21].因此,催化剂表面V=O 活性位点的数量直接影响VWTi 催化剂的脱硝和CB 降解活性.
为了查明催化剂上负载不同形态钾盐对催化剂表面V=O 活性位点数量的影响,对新鲜和中毒催化剂样品进行了FT-IR 表征分析,结果如图7所示.已有研究表明:吸附峰波长在1052 cm-1附近的吸收峰是催化剂活性组分V2O5中V=O 键的红外吸收峰[33].因此,新鲜催化剂吸收带在1048 cm-1附近处出现的吸收峰为V=O 的不对称伸缩振动峰.催化剂负载K2SO4、K2O、和KCl 中毒处理后,V=O 键的不对称伸缩振动峰强度较新鲜催化剂均有一定程度的减弱,说明催化剂表面V=O 活性位点数量减少,造成催化剂脱硝和CB 降解活性降低.以上结果也进一步证明了,中毒催化剂样品中K+与部分V=O 键发生了相互作用.
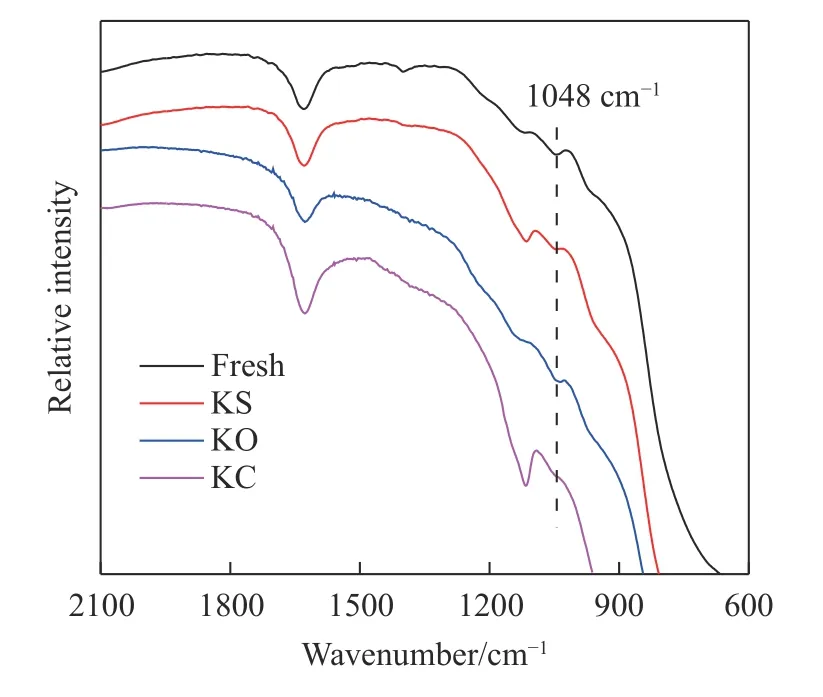
图7 新鲜与中毒催化剂的FT-IR 图谱Fig.7 FT-IR profiles of fresh and poisoned catalysts
2.1.7 对催化剂再生性能的影响
对三种钾盐中毒催化剂采用水洗和酸洗再生,对比研究了两种再生方式对中毒催化剂活性修复效果的影响,经水洗和酸洗再生后的催化剂分别记为:KC-W(水洗)、KO-W(水洗)、KS-W(水洗)、KC-A(酸洗)、KO-A(酸洗)、KS-A(酸洗).
对两种再生方式获得的催化剂样品进行了脱硝活性检测,结果如图8 所示.可以看出,水洗处理可以完全恢复催化剂的高温活性(350~450 ℃),而中低温(≤300 ℃)活性难以完全恢复.对于KC样品,在250 ℃时,其脱硝活性可以由中毒催化剂的26%提升到67%;在300 ℃时,水洗后催化剂脱硝活性由中毒催化剂的50%提升到81%.对于KO 样品,在250 ℃时,其脱硝活性可以由中毒催化剂的64%提升到74%,活性恢复效果非常有限;300 ℃时,水洗后活性也仅恢复到了新鲜催化剂脱硝活性的85%左右.相比之下,KS 样品采用水洗处理活性恢复效果最好,基本能恢复到新鲜催化剂的水平,250 ℃可以恢复到新鲜催化剂脱硝活性的93%.综合以上,不同形态的钾盐中毒催化剂,通过水洗工艺,可以有效的恢复其高温活性,但对中低温活性来说恢复效果不佳.

图8 再生催化剂脱硝活性检测Fig.8 Denitration activity of the regenerated catalyst
针对催化剂水洗中低温活性恢复效果有限的问题,采用质量分数1%的H2SO4酸洗再生,可以看出,三种钾盐中毒的催化剂,其活性并没有明显提升,甚至低于水洗再生结果.以受KCl 中毒的催化剂为例,300 ℃时,中毒催化剂脱硝活性为50%,采用水洗再生后其脱硝活性恢复到81%,而采用酸洗再生后其脱硝活性为78%.
对水洗和酸洗前后中毒催化剂样品进行XRF分析,结果如表4 所示,从表中可以看出,水洗对K+的去除率在84%~89%之间,而酸洗对K+的去除率都能达到100%,明显酸洗效果更佳.但同时,酸洗也会洗脱催化剂中部分V 元素.以KCl 中毒催化剂为例,水洗前后V 元素基本保持不变,酸洗再生后,V 元素的含量从新鲜催化剂的1.08%降低到0.75%,降低幅度达到了30%以上,这可能是采用酸洗再生并不能有效提高中毒催化剂活性的原因.

表4 不同形态钾盐中毒的催化剂再生前后XRF 分析结果Table 4 XRF results of the poisoning catalysts before and after regeneration
水洗和酸洗处理前后中毒催化剂样品的CB 降解活性检测结果如图9 所示,从图中可以看出,两种常规再生手段对中低温(≤300 ℃)条件下的CB 催化降解活性几乎没有恢复作用,高温(350~450 ℃)活性也仅有较小程度的恢复,整体而言,酸洗再生对中毒催化剂CB 降解活性恢复能力要强于水洗再生.虽然水洗和酸洗可以去除中毒催化剂表面的K+,但并没有较好的恢复中毒催化剂的氧化活性,可能是钾盐对催化剂表面产生了不可逆转的破坏,导致其氧化活性无法恢复.

图9 再生催化剂CB 降解活性检测Fig.9 CB degradation activity of the regenerated catalyst
2.2 不同形态钾盐致VWTi 催化剂失活机理分析
根据以上不同形态钾盐对催化剂活性及结构性能的影响研究,提出了不同形态钾盐致VWTi催化剂失活的机理,如图10 所示.K2SO4对催化剂的失活主要是K+会与催化剂表面的Brønsted 酸性位点(V—OH)和Lewis 酸性位点(V=O)反应生成V—O—K 键,导致活性位点对NH3和CB 的吸附能力下降,从而降低其催化还原和催化氧化活性,但SO42-的引入可以产生新的酸性位点(—OH基团),为NH3和CB 吸附提供新的位点参与脱硝和CB 氧化反应.对于K2O 致催化剂失活机理,我们认为,K+与催化剂表面的Brønsted 酸性位点和Lewis 酸性位点结合形成了V—O—K 键,导致催化剂表面活性中心对NH3和CB 的结合能力丧失,从而降低催化剂活性.对于KCl 致催化剂失活机理,我们认为,K+和Cl-都会与催化剂表面酸性位点反应,K+会与V—OH 反应形成V—O—K,Cl-会先与V=O 反应生成Cl—V—O—H,同时K+继续与Cl—V—O—H 反应生成Cl—V—O—K,使得催化剂活性降低.已有研究表明,反应过程中形成的Cl—键也可以表现出对NH3吸附能力,所以在中毒催化剂的NH3-TPD 实验中,KCl 中毒催化剂在Lewis 酸性位点表现出比K2O 中毒催化剂更强的吸附峰.但该吸附峰不具有活性,无法活化吸附NH3参与SCR 反应.同时,Cl—键也会造成催化剂出现Cl 中毒,减弱催化剂上CB 分子C—Cl键的亲核取代吸附作用.

图10 钾盐对VWTi 催化剂的失活机理.(a) K2SO4;(b) K2O;(c) KClFig.10 Deactivation mechanism of kali salts on the VWTi catalyst: (a) K2SO4 ;(b) K2O;(c) KCl
3 结论
(1)不同形态钾盐均会造成VWTi 催化剂的脱硝和CB 催化降解活性降低,对催化剂活性的影响顺序为:KCl> K2O> K2SO4;中毒催化剂水洗再生处理对其高温(>300 ℃)脱硝活性恢复效果明显优于中低温(≤300 ℃)脱硝活性恢复效果,但水洗和酸洗再生均无法恢复中毒催化剂的CB 降解活性,说明钾盐中毒对催化剂氧化性能产生了不可逆的影响.
(2)不同形态钾盐对催化剂的失活机理可以归纳为物理和化学中毒,物理中毒主要是毒物在催化剂表面沉积并堵塞孔道,但这不是催化剂活性降低的主要原因.而化学中毒主要是由于K+与催化剂表面的活性组分钒产生相互作用,钝化表面活性位点,进而降低了催化剂的脱硝和CB 降解活性.
(3)K+与催化剂表面氧形成了强键,占据表面化学吸附氧空位,从而使得催化剂表面活性物质氧化还原性减弱,表面酸性位点数量降低,表面中心上V5+比例和表面化学吸附氧的比例下降,这些是导致VWTi 催化剂化学失活的主要原因.
(4)催化剂中引入SO42-可以表现出Brønsted酸性质,形成酸性位点,可以吸附NH3并将其活化后参与SCR 脱硝反应;该酸性位点也可以吸附CB,并发生亲核取代反应,使得C—Cl 键断裂.
