金刚石/铜(银、碳化钛)界面性质的第一性原理计算
2022-11-04韩金江陈冰威路朋献栗正新
韩金江,陈冰威,路朋献,李 颖,栗正新
(河南工业大学 材料科学与工程学院,郑州 450001)
随着微电子技术的不断发展,电子器件不断向高功率密度、高集成度发展,但器件因严重的发热问题,使常处于高温下工作的电子器件的稳定性和寿命大大降低[1]。为解决其散热问题,各种高热导率材料成为该领域研究的热点,其中的金属基金刚石复合材料是重要的研究方向。常用的金属基体有铝、银和铜,但银和铜与金刚石之间的润湿性较差,通常采用金属基体的合金化[2-3]对金刚石表面进行处理[4-6]和采用合理的复合材料成形工艺[7-9]改善两者之间的界面结合。
目前已经有大量试验对界面结合改善情况进行了研究。LI 等[10]通过对金刚石铜表面镀钛得到了高热导率的金刚石/铜复合材料。WU 等[11]通过真空热压烧结法制备金刚石/铜复合材料,并引入钛改善其界面结合,与不添加钛元素的复合材料相比,其热导率提升了48%。JHONG 等[12]采用无压烧结法制备金刚石/银复合材料,并添加钛元素,制备出热导率高达953 W/(m·K)的复合材料,相当于其理论热导率的98%。TANG 等[13]通过在金刚石表面镀覆碳化铬来改善金刚石与银之间的界面结合,并优化了试验中的工艺参数,得到了热导率为695 W/(m·K)的金刚石/银复合材料。
另外,可以通过第一性原理揭示材料界面结合能力和热传导问题。CHEN 等[14]利用密度泛函理论和原子格林函数方法研究了金刚石/铜复合材料界面的纳米尺度的热运输问题,其声子谱的相关计算表明,金刚石与铜之间的声子耦合性较差,金刚石与铜之间的界面热阻较大。XIE 等[15]讨论了钼掺杂对金刚石(111)/Al(Cu)(111)界面相互作用、拉伸性能和热导率的影响,表明铝与金刚石之间的界面结合要优于铜与金刚石的,钼掺杂后对铝/金刚石界面的增强作用有限,但对于铜/金刚石界面的增强作用较大。
上述的计算和试验研究均取得了一定的成果,但对金刚石/银的界面结构和界面性质的研究尚未见报道,并且通过引入碳化钛来改善金属与金刚石之间的界面结合和热传导能力的机理尚未有相关解释。因此,采用第一性原理计算的方法研究金刚石/银、金刚石/铜、金刚石/碳化钛 3 种界面的结构、电子结构和传热行为。
1 计算方法与建模
1.1 计算方法
采用基于密度泛函理论的第一性原理方法,对3种界面结构的性质进行研究;采用Materials Studio 中的Materials Visualizer 模块进行模型构建; 采用CASTEP 模块进行结构优化[16]、能量计算以及界面性质的相关计算;采用FORCITE 模块进行径向分布函数的计算。采用平面波展开的赝势为超软赝势[17]。采用广义梯度近似(generalized gradient approximation,GGA)中的PBE(perdew-becke-ernerhof)[18]泛函对交换关联能进行处理。布里渊区中k点的选取方法为Monkhorst-Pack 网格[19]。收敛标准是系统总能量的变化在1×10-5eV 以内,结构优化后每个原子上的力小于0.03 eV/Å,晶胞的剩余应力小于0.05 GPa,公差偏移小于0.001 Å。在没有原子固定的情况下,采用BFGS(broyden-fletcher-goldfarb-shanno)[20]算法进行结构优化;在有原子固定的情况下,采用两点快速下降(two-point-steepestdescent,TPSD)[21]方法进行结构优化。结构优化和能量计算均采用的截断能为500 eV,k点网络为8×8×8;表面计算采用的截断能为400 eV,k点网络为8×8×1;界面模型最佳间距测试和结构优化采用的截断能为400 eV,k点网络为7×7×1。在处理表面结构和界面结构时,选取厚度为15 Å的真空层。电子态密度计算的k点网络为14×14×2。差分电荷密度是计算当前结构中每个原子及其孤立存在时的电子密度差异。
1.2 体相性质与表面性质
人造金刚石裸露的表面为(100)面和(111)面,而铜、银和碳化钛均属于立方晶系,面心立方布拉菲点阵,密排面均为{111}面。选用金刚石、铜、银和碳化钛的(111)面构建金刚石(111)/铜(111)、金刚石(111)/银(111)、金刚石(111)/碳化钛(111)的界面结构,揭示3 种复合材料的界面性质。
为使界面模型中各种物质的晶体结构符合现实情况,对采用的各种晶胞模型进行结构优化,将晶胞模型的晶胞参数(A)与其他人的计算结果(B)和试验数据(C)进行比对。铜、银、金刚石、碳化钛的晶胞参数见表1。由表1 可得:本文计算的晶胞参数满足计算所需要的精度要求。

表1 计算的铜、银、金刚石、碳化钛的晶胞参数以及他人试验和计算的结果Tab.1 Calculated cell parameters of copper, silver, diamond and titanium carbide, as well as the results of others’experiments and calculations
确定原子层数的方法为计算其表面结构的表面能。表面能计算的结果表明,6 层金刚石(111)面、6 层银(111)面、6 层铜(111)面的表面能趋于稳定。碳化钛原子层数的确定采用测量弛豫后原子层间距的方法,其表面原子弛豫的结果如表2所示,结果表明9 层碳化钛的内部原子层间距与块体碳化钛的原子层间距相似。因此,选择9 层碳化钛原子,并且以钛原子作为第1 层原子的表面结构构建其界面结构。

表2 碳化钛的表面原子弛豫Tab.2 Surface atomic relaxation of titanium carbide
1.3 界面的构建
由表1 可以看出:只有铜与金刚石的晶胞参数相近。因此,需要重新定义碳化钛和银的表面结构的向量,由u(1,0),v(0,1)改为m(1,-1),n(2,1)。铜与金刚石则进行扩胞使其变为原来的倍。之后,用式(1)计算金刚石/铜、金刚石/银、金刚石/碳化钛的3 种晶格失配度[30],分别为1.76%,0.52%,5.20%。经过表面重构之后,界面结构的失配度均较小,可以构成稳定的界面结构。界面处的几何参数以金刚石为基准,所构建的3 种界面模型和界面处的原子重叠方式如图1所示。

其中:Δ为晶格失配度,a1、a2分别为表面1 和表面2 晶格常数。
2 结果与讨论
2.1 界面间距和界面黏附功
采用通用结合能曲线(universal binding energy relation,UBER)测试图1 中3 种界面结构的界面间距,得到最为合理和稳定界面结构的UBER 曲线,如图2所示。图2 中的金刚石/铜黏附功最大时的界面结构间距最小,可能是在取界面间距时间隔过大,导致其数据有所偏差。

图1 3 种界面结构Fig.1 Three interface structures

图2 金刚石/铜、金刚石/银、金刚石/碳化钛界面结构的UBER 曲线Fig.2 UBER curves of interface structures of diamond/copper,diamond/silver and diamond/titanium carbide
黏附功计算公式如式(2)所示。在UBER 曲线中,黏附功从大到小依次为金刚石/碳化钛的界面、金刚石/银的界面、金刚石/铜的界面。
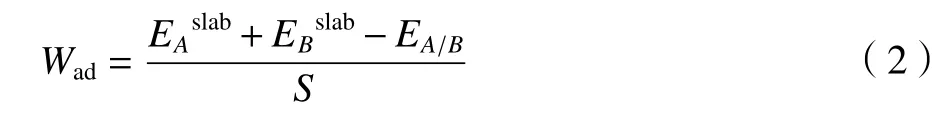
其中:EA/B为界面A/B的总能量,和分别表示构成界面A/B结构的表面结构的能量,S为界面结构的面积。
在固定非界面处原子的情况下,对于界面结构进行弛豫,并计算界面结构的黏附功,数据如表3所示。界面黏附功越大,界面间距越小,界面结合性能越强,界面结构越稳定。从表3 的界面间距的角度分析可知:金刚石/银(2.236 Å)的界面间距较大,而金刚石/铜(2.046 Å)和金刚石/碳化钛(1.990 Å)的界面间距相近。这表明,金刚石/银之间的界面结合性能较差。而从表3 的黏附功的角度分析,金刚石/碳化钛的界面黏附功(5.578 J/m2)相比于金刚石/铜的界面黏附功(1.919 J/m2)和金刚石/银的界面黏附功(4.291 J/m2)分别增加191% 和30%。这说明,金刚石/碳化钛的界面结合最强,结构最稳定。
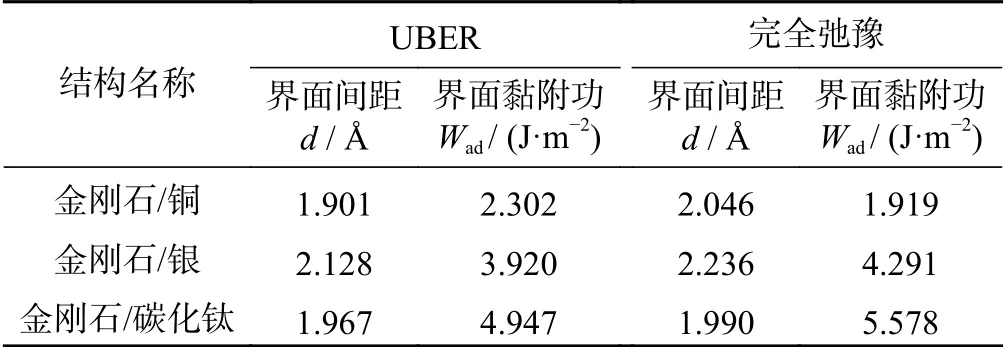
表3 通过UBER 和完全弛豫所得到的最佳界面间距和最佳黏附功Tab.3 Optimal interface spacing and adhesion work obtained by UBER and complete relaxation methods
2.2 电子态密度
图3 为电子态密度计算结果。在3 种界面结构中,界面附近的碳原子在费米能级处的态密度均不为0,即碳原子在金属原子的诱导下呈现出一定的金属性质。并且3 种金属原子均在-20 eV 附近出现新的峰值,表明金属原子与碳原子的轨道存在杂化作用。所有结构的第二层原子受界面结构的影响较小。
如图3a 和3b所示:在金刚石/铜界面结构中界面处2 种原子在-2 eV 附近存在杂化峰,金刚石/银界面结构中界面处2 种原子在-1 eV 附近存在杂化峰。2 个峰较为尖锐,表现出较强的局域化特性。但2 种结构中杂化峰数量较少,表明铜原子和银原子与碳原子之间相互作用较弱,即2 种界面结构的结合能力较低。

图3 3 种界面结构的电子态密度Fig.3 Electronic density of states of the three interface structures
如图3c所示:在金刚石/碳化钛的界面结构中,界面处钛原子和碳原子的波形发生了较大的变化。碳原子的态密度向费米能级移动,并且在-5~5 eV 处与钛原子的s 和p 轨道出现了多处共振峰和多处重叠区域。表面钛原子与碳原子之间相互作用较强,这种作用增大了界面结构的结合能力。
2.3 马利肯布居分析
3 种界面结构界面处原子轨道布居分析如表4、表5、表6所示。所取的原子为2 种表面结构中最靠近界面处的2 层原子,一共4 层原子。电荷的转移数越多,形成的键合作用越强。在金刚石/铜界面处,铜原子失去0.16 eV,碳原子得到0.20 eV。在金刚石/银界面处,银原子失去0.22 eV,碳原子得到0.16 eV。这2 种界面结构主要为金属原子的p 轨道失去电子,碳原子的s 和p 轨道得到电子。综合电子得失,金刚石/银界面键合作用更强。在金刚石/碳化钛的界面结构中,碳原子得到0.27 eV,s 和p 轨道均得到电子。这种结构的碳原子得到的电荷数目为3 种界面结构中最多,表明这种界面结构键合作用最强,界面结构最稳定。
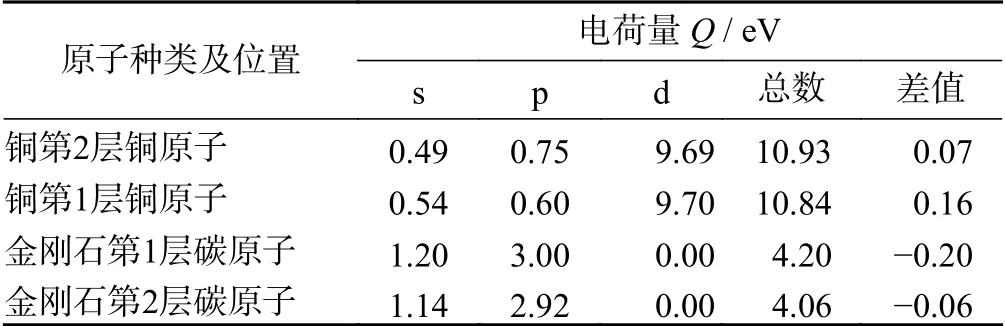
表4 金刚石/铜界面原子轨道布居分析Tab.4 Analysis of atomic orbital population at diamond/copper interface

表5 金刚石/银界面原子轨道布居分析Tab.5 Analysis of atomic orbital population atdiamond/silver interface

表6 金刚石/碳化钛界面原子轨道布居分析Tab.6 Atomic orbital population analysis of diamond/titanium carbide interface
2.4 差分电荷密度分析
界面结构的原子差分电荷密度,如图4a、图4b、图4c所示。在3 种结构中的金刚石部分,碳原子之间存在强烈的电荷增加,并呈现球形,显示出强共价键作用的特点。在金刚石/铜的界面处仅有极少部分的黄色区域,表示金刚石表面的原子仅得到极少部分的电荷,并且其黄色区域较为分散,没有固定的取向性。而在金刚石/银的界面处,黄色区域更加集中,并且位于银原子的下方,呈现出一定的方向性,这表面银原子与碳原子之间不仅存在一定量的电荷转移,并且有一定的键合作用。这种键合作用使得金刚石/银的界面结构更加稳定。而碳化钛与金刚石界面处的电荷转移则更为强烈,其颜色为红色并且更加集中,且位于钛原子的下方。这表明:钛原子与碳原子之间具有更多的电荷转移和更强的键合作用。
为了更清晰地表现这3 种界面结构的电荷转移和成键强弱,取金刚石表面正上方0.3 Å处的差分电荷密度图,如图4d、图4e、图4f所示。3 种截面处的电荷区域的形状均为哑铃形的截面,说明均为碳原子的p 轨道得到了电荷。图中颜色的深浅反映了电荷转移量的大小和成键的强弱,其中金刚石/铜界面结构为淡黄色,表现出极少的电荷转移和极弱的键合作用。而金刚石/碳化钛界面处则有红色部分出现,表现出强烈的电荷转移和强的键合作用。金刚石/银界面处的电荷转移情况则位于两者中间,即一般的电荷转移量和较强的键合作用。综上,金刚石/碳化钛界面结构电荷转移最多,成键作用明显,界面结构最稳定,界面结合最强。
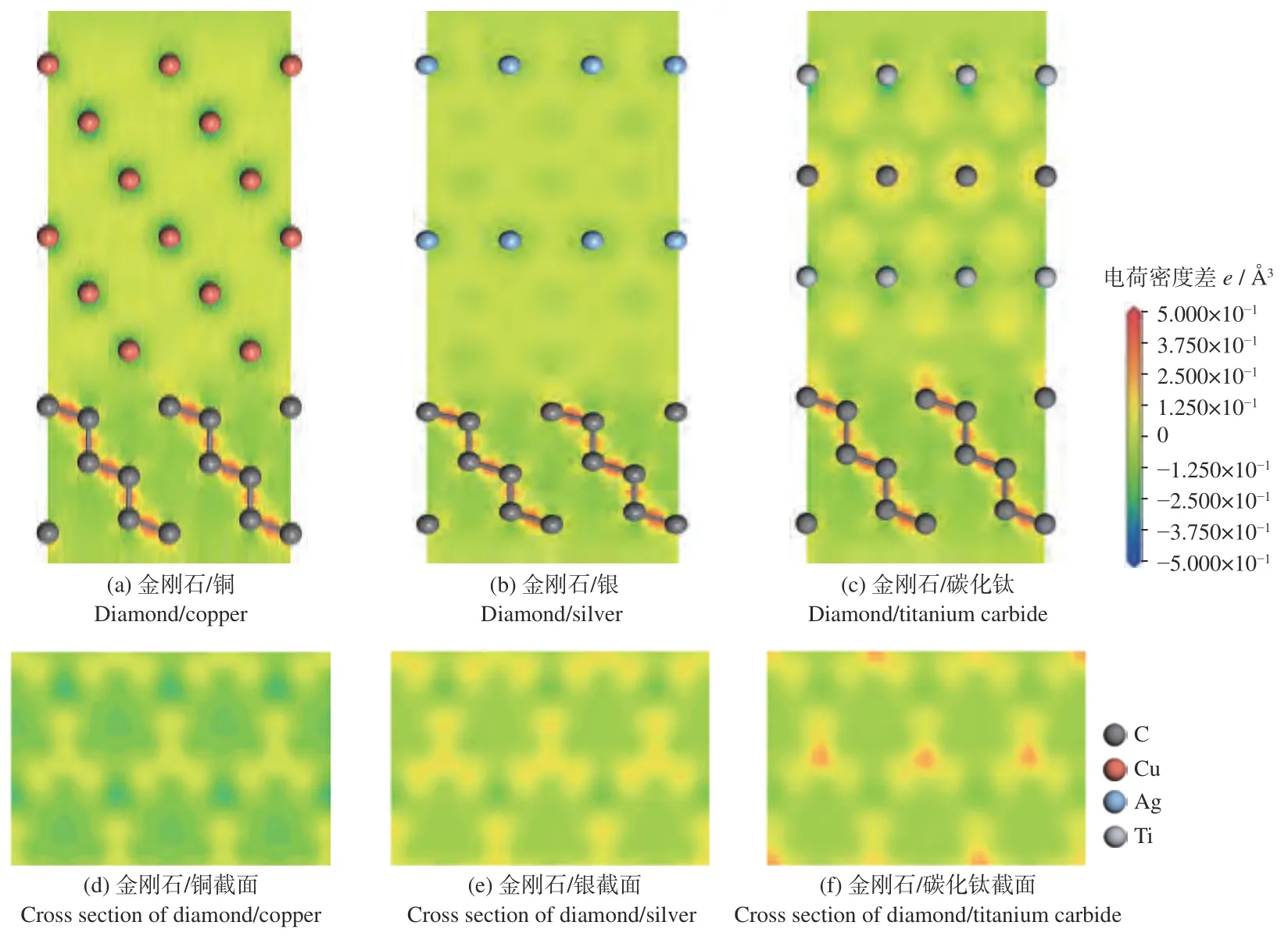
图4 3 种界面结构的差分电荷密度Fig.4 Three interface structure difference charge density
2.5 径向分布函数
3 种界面结构的径向分布函数如图5所示。所取的参考原子为金刚石表面结构的第1 层中处于中心位置的碳原子。只考虑5 Å以内的径向分布函数。在金刚石表面结构中碳原子与碳原子的距离有1.511 Å,2.493 Å,2.517 Å,2.923 Å,4.365 Å等,其对应的峰均在图5 出现。3 种界面结构的界面间距均在2 Å左右(见表3),在图5 中只有碳化钛与金刚石的界面结构在2 Å附近处出现了一个峰,表现出碳原子与钛原子存在相互作用,即两者表面的结合强度大。而在金刚石/铜与金刚石/银的图中并没有新的峰出现,表明碳原子与铜原子、碳原子与银原子间没有相互作用,即金刚石/银和金刚石/铜的结合强度较低。。

图5 径向分布函数图Fig.5 Radial distribution functions
2.6 声子态密度分析
金刚石、银、铜和碳化钛的声子态密度如图6所示。从图6 可以看出:金刚石声子的频率分布主要为15~40 THz,其峰值在37 THz 左右。而银与铜的声子频率分布与金刚石差距较大,这表明:金刚石与铜和银之间的声子耦合性能均较差,导致了金刚石与铜和银界面处的界面热阻较大,说明只采用金属和金刚石所制备的复合材料无法完全发挥金刚石热导率高的特点。而碳化钛的声子频率主要在0~13 THz 和15~20 THz。其中,较高的频率分布部分与金刚石的声子频率在15~21 THz,接近完全重合,这部分频率的声子存在使得金刚石与碳化钛之间存在一定的声子耦合性能,使其界面之间的热量传输能力有一定提高,从而提升了复合材料的热导率。

图6 声子态密度分布曲线Fig.6 Phonon density distribution curves
3 结论
(1)采用第一性原理计算分析了金刚石/银、金刚石/铜、金刚石/碳化钛的界面结构。相比于金刚石/铜和金刚石/银,金刚石/碳化钛的界面间距最小(1.990 Å),界面黏附功最大(5.578 J/m2),界面黏附功分别提高191%和30%。这说明金刚石/碳化钛的界面结合最强,界面结构最稳定。
(2)通过电子态密度,马利肯布居,差分电荷密度分析界面处的键合情况。金刚石/铜界面处存在电荷转移,但键合作用极弱。金刚石/银界面处存在键合作用,但强度不高。金刚石/碳化钛界面存在较多的电荷转移和较强的键合作用。
(3)通过计算声子态密度分析界面处声子耦合情况,结果表明:金刚石/铜和金刚石/银界面的声子耦合性均较差,金刚石与铜和银之间的界面热阻较大。金刚石/碳化钛界面之间声子耦合性较好,表明碳化钛的引入降低了界面热阻,提升了复合材料的热导率。
