在线固相萃取-超高效液相色谱-串联质谱法测定水体中氯胺酮、去甲氯胺酮和羟亚胺
2022-10-28朱思琪徐柏杨诸葛伟伟何颖声
朱思琪,徐柏杨,诸葛伟伟,何颖声
(国家毒品实验室浙江分中心(浙江省毒品技术中心)浙江省禁毒和毒情监测关键技术研究重点实验室,浙江 杭州 310053)
氯胺酮于1962年首次作为麻醉剂被合成,2003年我国公安部已将其列入毒品范畴。利用合法途径限制性使用氯胺酮是世界人民的共同期望,但目前依然有大量氯胺酮作为毒品被滥用。氯胺酮易导致精神分裂和致幻作用,不仅使吸食者生命健康受到损害,甚至会造成极其恶劣的社会危害,故应对其密切关注。去甲氯胺酮是氯胺酮在生物体内的代谢产物,在吸食氯胺酮人群的毛发、尿液及生活污水中均能检出。羟亚胺是氯胺酮的同分异构体,也是合成氯胺酮的前体,通过简单加热即可重排得到氯胺酮,并且无论通过哪一种合成路径,最终都是以羟亚胺作为氯胺酮合成的最终前体。2008年,羟亚胺作为一类易制毒化学品被严格管理。为了发现潜在的制毒工厂,排摸隐性吸毒人员,评价地区毒情形势,监测环境中的氯胺酮,有必要同时对氯胺酮、去甲氯胺酮及其前体羟亚胺进行有效监测。
污水流行病学(Wastewater-Based Epidemiology,WBE)基于对未经处理的废水中毒品及其代谢物含量的测定来统计区域毒品消费情况,成为毒品使用情况的有效监测手段。针对城市生活污水中毒品及其代谢物的研究较多,但是对于相关易制毒化学品的监测报道相对较少。羟亚胺的分析通常是基于气相色谱-质谱联用法(Gas Chromatography-Mass Spectrometry,GC-MS)、气相色谱法(Gas Chromatography,GC)或液相色谱-紫外光谱法(Liquid Chromatography-Ultra Violet,LC-UV)等分析方法。由于气相色谱仪进样口的温度及衬管的惰性程度均会影响其稳定性,难以避免一部分羟亚胺转变为氯胺酮,因此无法实现氯胺酮和羟亚胺的准确测定。LC-UV可满足氯胺酮和羟亚胺的同时准确测定,但其检出限仅为5 mg/L,难以满足实际水体中检测的需求。近年来,由于液相色谱-串联质谱联用法(Liquid Chromatography-Tandem Mass Spectrometry,LC-MS/MS)的测定灵敏度高,已广泛应用于污水中毒品及其代谢物的研究。周爽等首次建立了羟亚胺的LC-MS/MS分析方法,具有较好的稳定性和准确度,可用于氯胺酮和羟亚胺的同时测定,检出限可达1 μg/L。ZHANG等建立了离线固相萃取-液相色谱-串联质谱联用法(Solid Phase Extraction-Liquid Chromatography-Tandem Mass Spectrometry,SPE-LCMS/MS),实现了环境水样中甲基苯丙胺、苯丙胺、氯胺酮、麻黄碱和羟亚胺的同时测定,并对其发生、分布以及可能存在的环境危害进行了详细研究。
由于水样中氯胺酮及相关物质含量较低,且基质较为复杂,一般需要辅以样品前处理技术进行净化和富集,采用离线固相萃取技术过程较为繁琐,所需分析时间较长。为更好地对氯胺酮等相关物质进行快速高灵敏的分析,本文拟建立在线固相萃取-超高效液相色谱-串联质谱法(Solid Phase Extraction-Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry,SPE-UPLC-MS/MS)快速分析污水中氯胺酮及相关代谢物和制毒前体的新方法,并将其应用于浙江省大型污水处理厂和产业园区内污水样品的测定。
1 材料与方法
1.1 仪器与试剂
Waters ACQUITY Xevo TQ-XS型在线固相萃取液质联用仪(美国Waters公司)。
实验样品A~A来源于浙江省六个地级市的大型污水处理厂污水;实验样品B~B来源于浙江省产业园区污水。使用全自动采样器进行24 h混合样品采集,每个点位选择一个工作日和一个休息日,取样时间为2021年6—9月。
质量浓度为1 mg/mL的氯胺酮、去甲氯胺酮、羟亚胺标准品溶液,质量浓度为100 μg/mL的内标氯胺酮-D、去甲氯胺酮标准品溶液-D,(美国Cerilliant公司);甲醇、乙腈、甲酸均为色谱纯及以上;去离子水由Milli-Q超纯水系统制备。
1.2 方法
1.2.1 仪器工作条件
在线固相萃取条件Waters Oasis HLB固相萃取柱(2.1 mm×30 mm,20 μm);进样量为500 μL;流动相A为水,流动相B为乙腈,四元泵用于在线固相萃取,具体条件见表1。

表1 四元泵工作条件
色谱条件Waters ACQUITY UPLC HSS T色谱柱(2.1 mm×100 mm,1.8 μm);柱温40℃;流动相A为0.1 %(体积分数,下同)甲酸水溶液,流动相B为乙腈,二元泵梯度洗脱程序见表2。
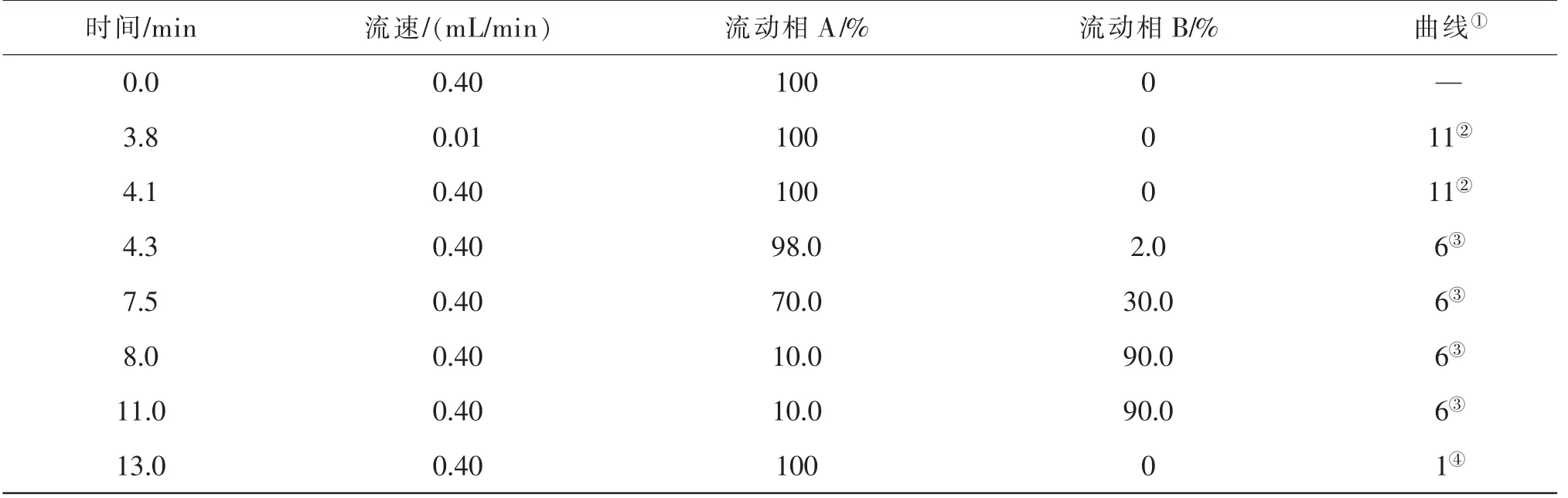
表2 二元泵梯度洗脱程序
质谱条件 电喷雾离子源(Electron Spray Ionization,ESI),正离子模式(ESI),多反应监测(Multiple Reaction Monitoring,MRM)模式;离子源温度600℃;喷雾电压0.5 kV;各目标物的MRM优化参数见表3。
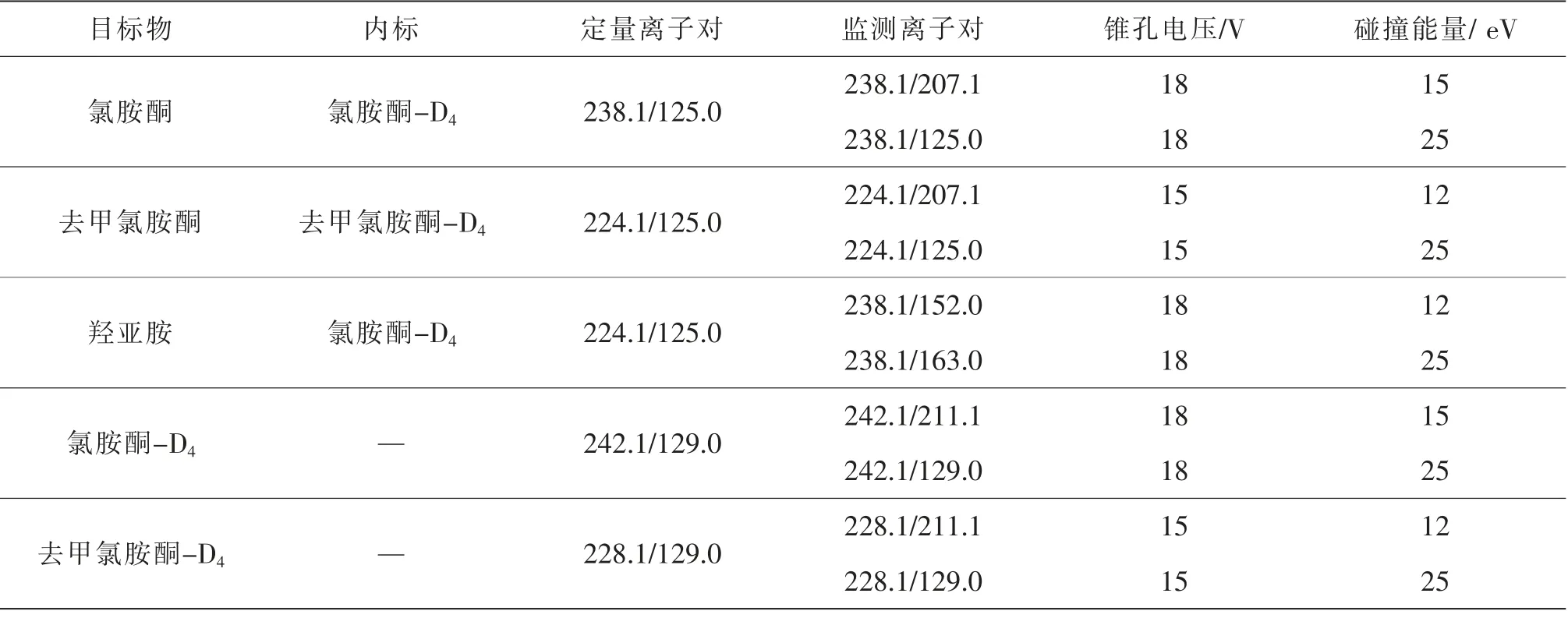
表3 各目标物的MRM优化参数
1.2.2 样品处理过程
取样品经0.45 μm滤膜过滤,取滤液50 mL转移至离心管中,加入质量浓度为25 ng/mL的氘代内标混合溶液100 μL,涡旋混匀,用0.22 μm滤膜过滤后待测。每份样品平行操作两次,空白溶液取超纯水50 mL与样品同法操作。
1.3 方法学验证
1.3.1 选择性
取超纯水,按已建立的方法分析考察空白水样对目标物及内标是否存在干扰。
1.3.2 标准曲线、检出限与定量限
按照实验方法对各质量浓度的标准溶液进行测定,考察方法线性。以目标物和相应内标物峰面积的比值作为横坐标(x),以目标物的质量浓度为纵坐标(y)绘制标准曲线,得到线性回归方程及相关系数。根据国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)规定,以3倍信噪比、10倍信噪比计算检出限与定量限。
1.3.3 精密度与准确度
取自来水,分别添加不同量的混合标准储备溶液,得到目标物质量浓度分别为2、50、150 ng/L的溶液,每个质量浓度平行测定6次,计算回收率,并以测定值的相对标准偏差作为日内精密度,连续测定3 d,计算日间精密度。
1.3.4 基质效应
分别对自来水、超纯水加标至质量浓度为2、50、150 ng/L,采用公式(1)计算基质效应。
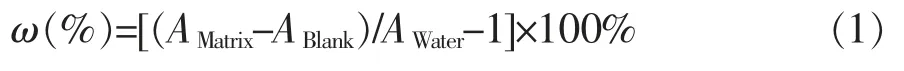
其中:A为目标物在基质中定量离子对的峰面积;A为基质空白中定量离子对的峰面积;A为目标物在超纯水中定量离子对的峰面积。
1.3.5 稳定性
取50 ng/L加标样品,分别放置0、24、48 h后进样测定,以考察样品的稳定性。
2 结果与讨论
2.1 色谱行为
分别取质量浓度均为50 ng/L的氯胺酮、去甲氯胺酮和羟亚胺单一标准储备溶液、质量浓度均为50 ng/L的氯胺酮-D、去甲氯胺酮-D内标溶液和空白水样进样。结果表明,3种目标物的分离情况良好,保留时间分别为7.88、7.72、8.23 min,基质对其他物质的定性、定量均无干扰。目标物的提取离子色谱图如图1所示。

图1 目标物的提取离子色谱图
2.2 标准曲线、检出限与定量限
3种目标物的线性参数、检出限与定量限结果如表4所示。本方法对3种目标物的检测范围均为0.2~200 ng/L,检出限为0.02~0.05 ng/L,定量限为0.05~0.1 ng/L,相关系数均大于0.999,可满足实际检测的需求。

表4 线性参数、检出限与定量限
2.3 精密度与准确度实验
本方法对氯胺酮、去甲氯胺酮和羟亚胺的平均加标回收率分别为98.0%~101%、99.3%~100%、91.3%~102%,3种目标物在不同加标质量浓度下的日内精密度和日间精密度为0.6%~3.5%,具体结果见表5。
2.4 基质效应
结果如表5所示,3种目标物的基质效应为99.8%~114%,通过内标校正基本可消除基质效应的影响。

表5 精密度与回收率实验结果
2.5 稳定性
实验对质量浓度为50 ng/L加标样品的稳定性进行考察。结果发现,氯胺酮和羟亚胺质量浓度的变化仅有0.7%、0.8%,去甲氯胺酮的质量浓度下降了9.0%,这表明样品在48 h内基本稳定。
2.6 实际样品分析
按照实验方法对所有大型污水处理厂和产业园区污水样品进行测定。结果表明:仅有样品A、A、A和B中检出氯胺酮原体,质量浓度分别为0.45、0.36、1.57、0.43 ng/L;所有样品均未检出去甲氯胺酮和制毒前体羟亚胺。
2.7 方法比较
目前尚未有文献报道同时分析水体中氯胺酮、去甲氯胺酮和羟亚胺的方法。将本文方法与已报道的文献相比,结果见表6。由此可知,本方法具有较低的检出限,无需离线活化固相萃取柱、真空干燥萃取柱、氮吹等步骤,显著减少了前处理步骤及分析时间。

表6 不同分析方法的性能比较
3 结论
本文基于在线SPE-UPLC-MS/MS技术建立了同时快速分析水体中氯胺酮、去甲氯胺酮和羟亚胺的新方法。该方法具有较低的检出限和较宽的线性范围,且前处理过程较为简单,仅需要13min即可同时实现水体中痕量氯胺酮、去甲氯胺酮和羟亚胺的测定,可成功应用于实际污水样品的分析,为分析水体中氯胺酮及其代谢物和制毒前体提供新的监测方法。
