基于非依赖数据采集的呼出气冷凝液蛋白质组加权基因共表达网络分析
2022-10-25孙东晓镇华君修光利
马 琳, 孙东晓, 镇华君, 修光利
(1. 华东理工大学国家环境保护化工过程环境风险评价与控制重点实验室, 上海 200237;2. 宾夕法尼亚州立大学医学院质谱中心, 美国 PA 17033)
呼出气冷凝液(Exhaled Breath Condensate,EBC)是一种来自于下呼吸道的衬液,常被用作肺部疾病研究的载体,尤其是EBC 的蛋白组学更是国内外研究热点[1]。EBC 的收集过程简便、无创,携带着大量的生理信息,理想情况下可以通过研究EBC 的蛋白组成来探究肺癌等相关肺部疾病的内在生物学特征,有利于提高对疾病的认知,并助益于疾病的诊察[2-3]。
随着质谱仪器的更新发展,蛋白组学技术也相应得到提高,进一步促进了蛋白质组在生物标志物方面的应用。以往研究中对EBC 蛋白组学的探索虽然从未间断,成果却十分有限,这主要是因为EBC 中极其微量的蛋白浓度无法使用一般的质谱方法进行解析,尤其是对于大量低丰度蛋白,往往会因高丰度蛋白的掩盖而被忽略,这严重限制了EBC 蛋白组学的发展[4]。绝大多数研究都是用液相色谱-串联质谱联用仪(LC-MS/MS)来进行EBC 蛋白组学研究,在LC-MS/MS 分析中,数据采集策略会对鉴定结果造成显著的影响[3]。数据相关采集(Data-Dependent Acquisition,DDA)是常用的采集策略之一,但DDA的采集策略是有偏倚的,对前体信号强的碎片进行选择性捕获,对于信号较弱的离子捕获性不强[4]。数据独立采集(Data-Independent Acquisition,DIA)是一种最新发展的数据采集技术,不同于DDA 的信号捕集策略,DIA 会对所有的离子信号进行捕获,在二级质谱(MS2)扫描阶段进行全窗口的扫描,对信号强度没有依赖,这样得到的信息避免了选择性缺失,并高度可重复[5]。将DIA 方法运用到EBC 蛋白组学研究中,可以大大提高蛋白质的鉴定水平,是研究EBC 蛋白组学的理想工具。
加权基因共表达网络分析(Weighted Gene Coexpression Network Analysis,WGCNA),是在传统生物信息学分析上衍生出的多维分析[6]。WGCNA 是一种全新的算法,其逻辑在于蛋白质是无尺度分布的,不是单独的个体,而是以组群的方式存在,根据不同的表达模式,划分为不同的模块(module)。在生物学分析的过程中,以模块为单位进行聚类分析,将不同模块之间的关系、不同性状之间的关系连结起来,来筛选重要模块蛋白,这些模块蛋白往往具有最显著的生物功能,在疾病研究中用来筛选生物标记物和治疗靶点[7]。
目前,国内外鲜见使用DIA 方法进行EBC 蛋白组学分析,并使用WGCNA 算法分析其生物功能的研究。本文使用DIA 分析EBC 蛋白组成分,在此基础 上 结 合WGCNA 和Gene Ontology(GO)分 析、Kyoto Encyclopedia of Genes and Genomes(KEGG)分析、Protein-Protein Interactions(PPIs),探讨了WGCNA结合蛋白组学在实际应用中的价值。
1 实验部分
1.1 主要材料与试剂
胰蛋白酶(Trypsin)、二硫苏糖醇(dithiothreitol,DTT)、碘乙酰胺(iodoacetamide ,IAA)、尿素(Urea ,UA)、三羟甲基氨基甲烷盐酸盐(Tris-HCl)购自西格玛奥德里奇(上海)贸易有限公司;iRT 标准肽段购自瑞士Biognosys 公司;甲酸购自阿拉丁试剂(上海)有限公司;10 kDa 超滤膜购自默克密理博实验室设备(上海)有限公司;RTube 购自美国Respiratory Research Inc 公司。
1.2 纳入人群
本研究共纳入30 名受试者,包括10 名肺癌患者(Lung Cancer,LC)、10 名 良 性 肺 部 疾 病 患 者(Pulmonary Nodules,PN)和10 名健康对照(Healthy Controls,H)。样本于2018 年4 月至2018 年7 月在上海胸科医院采集,所有EBC 样本在上午8 点至9 点采集完毕,并立即存储于超低温冰箱,以备后用。呼出气采集装置为RTube,全程佩戴鼻夹。
1.3 样品制备
使用胰蛋白酶进行蛋白酶解,结合超滤辅助的样品制备方法(FASP)进行EBC 样本的过滤和浓缩。每个样品用10 kDa 滤膜浓缩,与100 μL UA 缓冲液(8 mol/L 尿素,150 mmol/L Tris-HCl,pH 8.0)和DTT混合至10 mmol/L 的最终浓度,然后与100 μL IAA(50 mmol/L IAA in UA)、100 μL NH4HCO3缓冲液(50 mmol/L)和40 μL NH4HCO3缓冲液(0.5 μg 胞内蛋白酶Lys-C)混合。然后,向样品中添加0.5 μg 胰蛋白酶进行过夜酶解,并与40 μL NH4HCO3缓冲液(50 mmol/L)混合。然后将样品以14 000 倍重力加速度离心浓缩30 min,收集滤液并冷冻干燥。每个样品用12 μL、φ=0.1% 甲酸(FA)复溶,280 nm 波长处吸收波段(OD280)用于测量肽浓度。然后,从每个样品中提取5 μL 肽段(约0.5 μg),并混合2 μL iRT 标准肽段用于质谱分析。
1.4 质谱分析
质谱分析分为两个步骤,DDA 分析和DIA 分析。DDA 分析中,使用EASY nLC 1200 系统(Thermo Fisher Scientific,CA)和C18 柱(75 μm×300 mm,3 μm)进行色谱分离。缓冲液A 为φ=0.1%甲酸水溶液,缓冲液B 为φ=0.1% 甲酸乙腈水溶液(乙腈体积分数为 84%)。使用2 h 线性梯度,流速为250 nL/min,以φ=95%缓冲液A 平衡:梯度8%~30% 持续97 min,30%~100%持续13 min,并保持10 min。分离后,通过Q-Extractive HF 质 谱 仪(Thermo Fisher Scientific,CA)进 行DDA 分 析。扫 描 范 围(m/z):300~1 800;质 谱 分 辨 率:60 000;AGC (Automatic gain control):3×106;Maximum IT:50 ms。MS 扫 描 后 继 续 进行20 个MS2 扫描,Isolation window:1.6 Th;质谱分辨率:30 000;AGC :3×106; Maximum IT: 120 ms;MS2 Activation Type:HCD;标准化碰撞能量:27。
DIA 分析与DDA 分析使用的系统相同。梯度分离条件为:梯度10%~30%持续97 min,30%~100%持续13 min,并保持在100% 直到120 min。DIA 扫描范围(m/z):350~1 650,分辨率:120 000,AGC:3×106,Maximum IT:50 ms。设置30 个DIA 窗口进行MS2 扫描。对于MS2 扫描,分辨率:30 000,AGC:3×106,Maximum IT “自动”,碰撞能量:25,光谱数据类型:“profile”。
1.5 数据分析
使用Spectronaut pulsar X 软件进行蛋白质鉴定。WGCNA 分析使用R 软件(Version 6.4)数据包完成。GO 分析使用Blast2GO 完成,KEGG 分析通过KAAS(KEGG Automatic Annotation Server)完成。
2 结果与讨论
2.1 蛋白质鉴定结果
所有酶解样本的样品经DDA 和DIA 质谱数据采集后,使用Spectronaut pulsar X 软件构建蛋白库。蛋白库由两部分组成,一部分为DDA 定量数据,另一部分为DIA 数据在pulsar 中直接检索后构建的Library。共鉴定到蛋白质2 052 个,其中肺癌组866 个,肺结节组1 129 个,健康对照组1 089 个。大部分蛋白质没有在此前的研究中报道过,是目前为止最全面的EBC 蛋白谱[8]。研究表明,基于DIA 的组学方法可以有效开发EBC 蛋白成分,提高了EBC蛋白质组学的敏感性和特异性。
利用DIA 技术建立了EBC 的蛋白组学方法,克服了EBC 样本蛋白浓度过低、常规方法无法完成蛋白组学研究的困难。基于DIA 的蛋白质组方法,对EBC 中低丰度蛋白有很好的鉴定能力。使用超滤管酶解FASP 的样本制备方法,不仅可以将样本过滤浓缩,还可以去除高分子聚合物的影响,这些聚合物来自于EBC 收集管,不可避免地干扰到蛋白质的鉴定。在以往的研究中,对EBC 的处理往往是简单的冷冻浓缩,并没有考虑到样本污染问题,因此往往不能取得令人满意的蛋白鉴定结果[9]。
此前,已有研究人员使用LC-MS/MS 方法对EBC 的蛋白组学进行探索,然而由于技术的限制,这些研究并没有很好地挖掘出EBC 的蛋白成分,也不能进行更深入的生物信息学分析[10]。Muccilli 等[3]对9 例EBC 样本进行分析,共鉴定167 个蛋白;Sun等[11]用TMTs (Tandem Mass Tags)方法对38例EBC 样本进行了蛋白组学分析,鉴定到257 个蛋白,之后对两组蛋白进行差异蛋白分析,共发现24 个显著差异表达的蛋白,生物信息学分析结果表明,这些蛋白在COPD 疾病进展中起着至关重要的作用,表明EBC的蛋白质组学分析可用于相关疾病生物标志物的鉴定。国内外对EBC 蛋白组学的研究一直在持续探索,然而结果却并不令人满意,方法的灵敏度是限制EBC 蛋白组学研究的主要原因之一。DIA 方法对低浓度的样本展现出了极高的灵敏度,已有研究[12]表明30 min 的DIA 分析相当于120 min 的DDA 分析,能够鉴定两倍以上的肽段,蛋白质鉴定也相对提高25%。
2.2 WGCNA 分析
WGCNA 是无尺度分布的拓扑网络分析,这对研究蛋白质的互作关系十分有利,据此可以构建大型的蛋白网络,以此来观察蛋白质之间的关系,并筛选出表达模式相近的模块蛋白[13]。WGCNA 在蛋白质组中的分析步骤主要为蛋白表达、网络构建、模块分析和模块-性状分析和关键蛋白分析。网络构建的节点是蛋白表达,蛋白质之间的相关性是模块分析的依据。在进行WGCNA 分析时,需要选择软阈值,以此来确定网络是否符合无尺度分布。软阈值的选择通常为相关系数R2>0.8,并保证一定的连续性[14]。本文使用Pick Soft Threshold 函数自动筛选软阈值,如图1 所示。

图1 最佳软阈值筛选:(a)基于R2=0.9 无尺度网络的软阈值筛选;(b)软阈值为5 时网络的连通性Fig.1 Screening of the best soft threshold: (a) Soft threshold of scale-free network based on R2=0.9; (b) Connectivity when the soft threshold was 5
以5 为软阈值构建蛋白模块的聚类树图,如图2所示。图中每个颜色代表一个蛋白模块,灰色代表无法被分类的蛋白,分枝的远近代表蛋白的相似程度。通过蛋白聚类树可以看出,本研究的蛋白有很强的模块性。

图2 EBC 蛋白共表达模块划分Fig.2 Cluster dendrogram and module overview for EBC proteome
利用Topological Overlap Matrix(TOM)对所有蛋白进行聚类热图分析,如图3 所示。图中表达模式相近的蛋白被分类到同一个分支中,热图颜色越深,代表蛋白之间的重叠程度越高,蛋白质之间的功能越密切。

图3 基于TOM 的拓扑网络热图Fig.3 Heatmap of the topological network based on TOM
模块与性状之间通过相关系数表现其关联,通过计算相关系数和p值,可以筛选出与表型性状显著关联的共表达模块。图4 是模块与表型性状相关性热图,左侧纵坐标代表不同的模块类型,中间色块代表蛋白模块,根据色块上的相关系数和括号中的p值可以筛选出最显著的蛋白模块,图例代表相关系数R2的大小范围为-1.0~1.0,其中红色代表正相关,蓝色代表负相关。
根据上述结果,将共表达模块中的蛋白在所有样本中的表达进行聚类热图分析,可以看出每个模块在样本中的特征值分布,如图5 所示。图的上半部分为蛋白在各个样本中的表达模式热图,红色代表上调表达,绿色代表下调表达。下半部分蓝色模块为特征值的分布,绝对值越大代表样本整体表达变化量越大。

图5 蛋白表达热图(a)及蓝色模块特征值分布图(b)Fig.5 Heatmap of the eigenproteins expression (a) and module eigenvalue
通过观察模块特征值的聚类树图和聚类热图,可以筛选出与表达模式最相似的模块。由图4 可知,蓝色模块的相关性系数为 0.420,p值为 0.02,是4 个模块中表达最为显著的蛋白模块,这表明该模块中的蛋白可能共同参与了某些生物过程,协同发挥重要的生物功能,有挖掘生物标志物的潜力。以此分析蛋白重要性与模块关系的关系,探究蛋白与模块的相关性和蛋白与性状的相关性是否有良好的一致性,筛选可能承担最多生物功能的关键蛋白(Hub Protein),如图6 所示。

图4 模块与表型性状相关性热图Fig.4 Heatmap of the module-trait correlations

图6 蛋白重要性与模块关系的散点分布图Fig.6 Scatter plot of protein significance and module membership
经过上述分析,共有61 个蛋白被筛选为关键蛋白。对关键蛋白进行GO 和KEGG 分析,结果分别如图7 和图8 所示。GO 分析结果表明,EBC 关键模块蛋白的生物过程主要集中在磷代谢相关过程和细胞活动;KEGG 分析结果表明,这些关键模块的蛋白较多参与了人类疾病分类的代谢活动,在人体免疫活动和信号传导过程中也十分活跃。
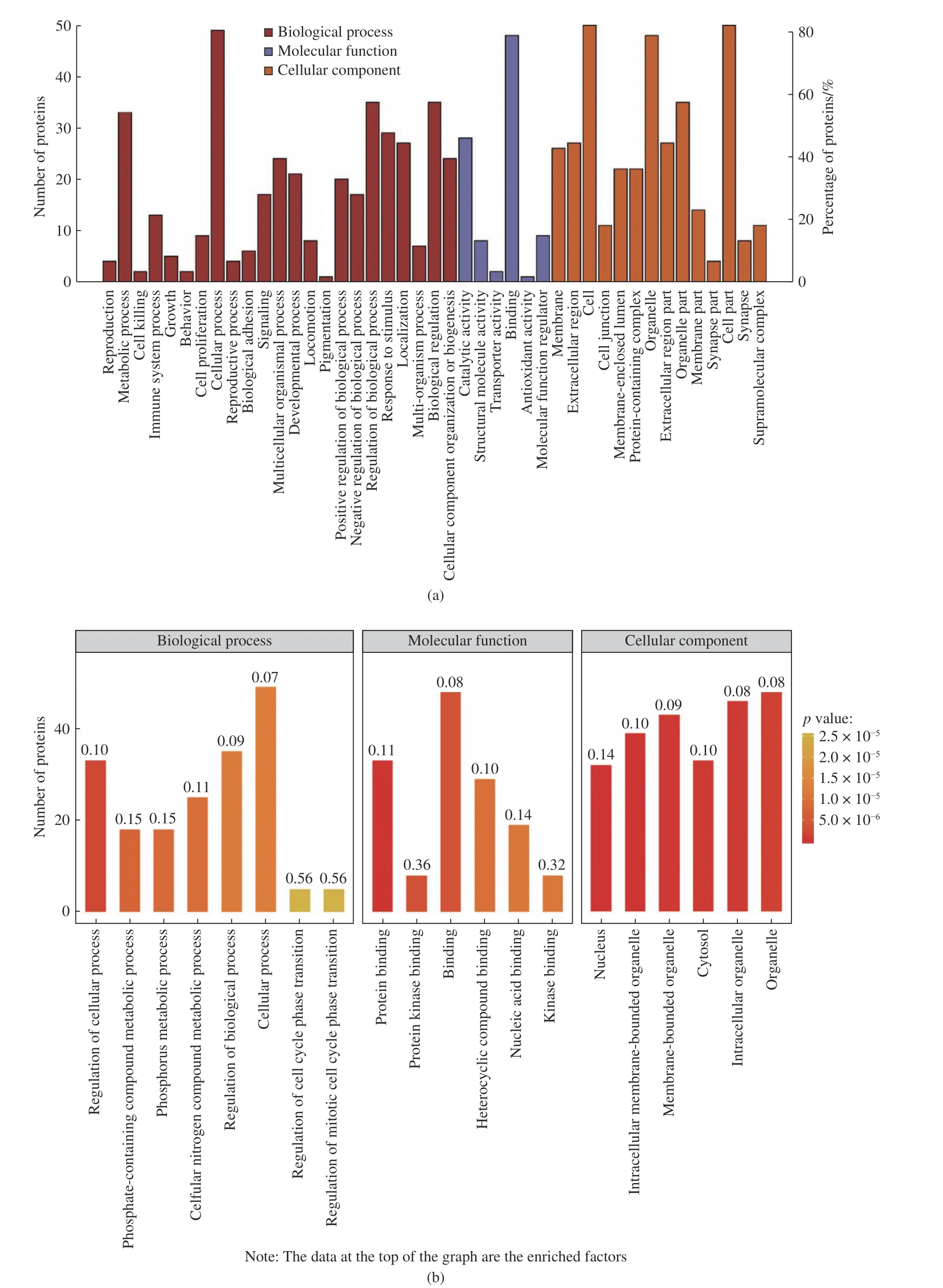
图7 关键模块蛋白的GO 分析: (a) GO term 分类;(b) GO 富集分析Fig.7 GO analysis of proteins extracted from the core module: (a) GO terms classification; (b) Enriched GO terms

图8 关键模块蛋白的KEGG 分析: (a) KEGG term 分类;(b) KEGG 富集分析Fig.8 KEGG analysis of proteins extracted from the core module: (a) KEGG terms classification; (b) Enriched KEGG terms
对一些关键蛋白的互作关系进行分析和可视化,结果如图9 所示。在String 中导入61 个关键蛋白,将结果展示设置为最高置信度(0.900),隐藏不产生连结关系的蛋白。图9 中的每一个节点都代表一个蛋白,蛋白与蛋白之间的连结线越多,说明蛋白之间的互作关系越大。结果表明,PPIs(Protein-Protein Interactions)网络共52 个节点和43 条连结线,平均节点为0.453,p<0.001。其中ACTB,HSPA8,TUBA4A,MDH2,HSP90AA1 等处于互作网络的核心区域,有可能是承担最多生物学功能的蛋白。
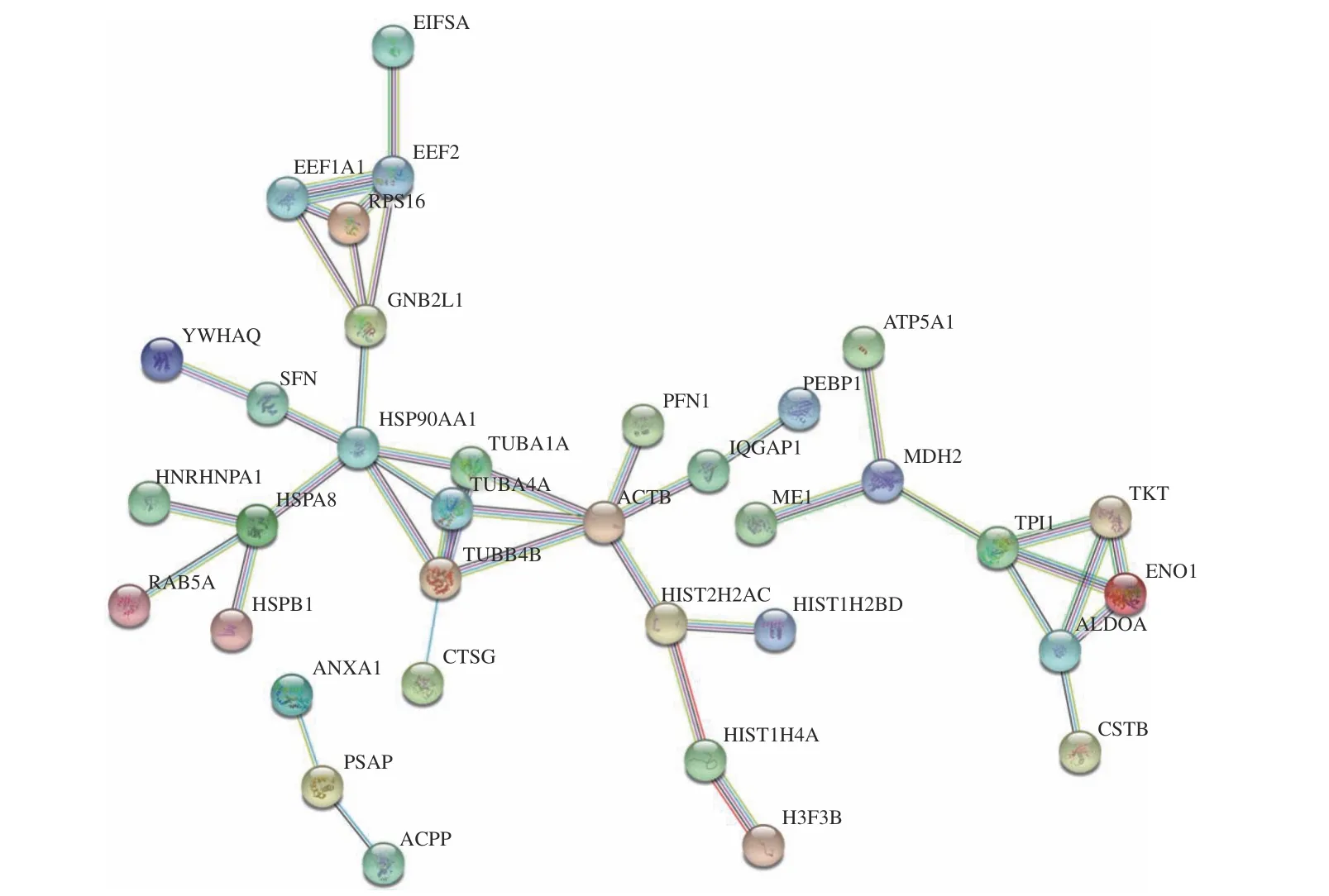
图9 蛋白互作网络分析Fig.9 Protein-protein interactions analysis
2.3 讨论
本文在EBC 蛋白组学方法建立的基础上,对鉴定到的蛋白进行了多维的生物信息学分析,对肺癌、肺结节和健康人群的蛋白组的组成和生物学功能有了初步的了解。WGCNA 网络对于处理无尺度分布的蛋白质组数据有天然的优势,本文依据蛋白间相互作用关系来挖掘蛋白内在关系,进一步筛选具有相似生物学功能的关键模块和关键蛋白。传统的生物信息学分析虽然对高表达的蛋白和基因有很强的分析能力,但对于低表达的蛋白功能挖掘能力较弱。WGCNA 分析可以把表达模式相似的蛋白归于同一个网络,将复杂的蛋白组学数据转化为不同网络和功能模块,再进一步分析每一个模块的功能,对处理大批量的蛋白组学数据有很大的优势[15]。
本文利用DIA 技术建立了基于EBC 的蛋白质组学分析方法。对30 例EBC 样本的分析共鉴定出蛋白质 2 052 个,其中肺癌组 866 个,肺结节组 1 129个,健康对照组1 089 个,表明基于DIA 的蛋白组学方法对低蛋白浓度的生物样本有很强的分析能力。使用WGCNA 算法,对EBC 样本的蛋白质组进行了分析,筛选出EBC 中发挥重要生物功能的核心蛋白。通过GO 分析发现这些关键蛋白在细胞核和细胞质中广泛存在,在分子功能方面,大部分互作蛋白发挥了结合功能,包括蛋白质结合、激酶结合和杂环化合物结合等。此外,这些蛋白质在细胞过程调节、磷化合物代谢、细胞含氮化合物代谢和有丝分裂周期等生物活动中十分活跃。KEGG 分析则提示关键蛋白参与了与免疫相关的系统性疾病、病毒致癌、细胞凋亡、Rap1 信号通路等代谢活动,这表明关键蛋白不仅活跃参与了与人体疾病相关的代谢活动,还参与了细胞的生理过程和信号传导,广泛涉及系统性疾病、肿瘤、感染类疾病等通路。
以上研究表明,WGCNA 所筛选出的模块蛋白具有生物学意义,能够反映EBC 蛋白组的生物功能,结合DIA 蛋白组学方法,可以开展更大规模的研究,在未来对肺癌等肺部疾病的研究和探索有很强的实际应用价值,可以助益生物标志物的探索和疾病诊疗。
