重组人瘦素蛋白的原核表达、纯化和生物学活性鉴定
2022-10-20周向阳李仲凯郝腾飞丁光硕郑文慧李文娟
周向阳,李仲凯,郝腾飞,丁光硕,郑文慧,李文娟
(1.河北大学 基础医学院,河北 保定 071000;2.河北省炎性自身免疫性疾病发病机制及防治重点实验室,河北 保定 071000;3.河北农业大学 生命科学学院,河北 保定 071001;4.河北大学 公共卫生学院,河北 保定 071000)
瘦素(Leptin)是由定位于人类染色体7q31.3的肥胖基因(Obese gene,Ob)表达的产物,是167个氨基酸组成的非糖基化蛋白[1].成熟的Leptin是切掉N端21个氨基酸信号肽后的具有强亲水性的单链球形分子,分泌入血后,以游离状态和结合状态2种形式存在,前者为生物活性状态[2].Leptin作为脂肪组织和中枢神经系统联系的外周信号,多数研究集中于其在体质量调节方面的作用,旨在阐明人类肥胖的机理[3].然而,越来越多的数据表明,Leptin不仅在调节食物摄入和能量平衡方面发挥着重要作用,而且还作为一种神经内分泌激素调节机体多种生理功能[4-5].研究表明,Leptin与肥胖、糖尿病、心血管等疾病的发生密切相关[6];同时,肥胖患者血浆中高水平的Leptin在多种肿瘤的发生和转移过程中发挥重要作用[7].
根据Leptin的非糖基化蛋白特性,具有生物活性的hLeptin可以在原核生物中有效表达.因此,研究者试图在大肠杆菌中表达重组hLeptin,但大多数重组hLeptin主要表达于不溶性包涵体[8].已有研究从大肠杆菌包涵体中纯化重组hLeptin,然而,对于商业规模生产而言,程序复杂而且效率低下[9].此外,瘦素的生物学效力根据所采用的重组表达折叠方法不同也有很大差异[10].
本研究主要通过大肠杆菌(Escherichiacoli)的密码子使用偏向性合成了hLeptin成熟肽编码序列,并在大肠杆菌中以SUMO融合标签的形式高效表达重组hLeptin,生物学活性实验表明,纯化的hLeptin具有完整的生物活性,这些都为进一步了解hLeptin的生物学功能和生理作用机制奠定了基础.
1 材料与方法
1.1 材料
1.1.1 质粒与细胞
大肠杆菌 pET-30a(+)载体购自Invitrogen公司.大肠杆菌DH5α和BL21(DE3)感受态细胞均购自北京博迈德基因技术有限公司.人肝癌HepG2细胞由本实验室保存,细胞生长于体积分数为10%的胎牛血清、2 mmol/L谷氨酰胺的DMEM培养基,于体积分数为5% CO2的细胞培养箱中37 ℃培养.
1.1.2 试剂
FastPfu Fly DNA Polymerase购自TransGen Biotech公司;2×Seamless Cloning Mix、琼脂糖均购自Biomed公司;质粒提取试剂盒购自Biomiga公司;胶回收试剂盒购自Promega公司;BCA法蛋白浓度测定试剂盒购自Sangon Biotech;蛋白胶试剂盒购自康维世纪生物科技有限公司;0.01 mol/L PBS溶液(磷酸盐缓冲溶液)、CCK8试剂购自Solarbio公司.
1.1.3 实验动物
6~8周龄的健康雄性KM小鼠12只,体质量(25.3±1.2) g,购自北京维通利华实验动物技术有限公司.按照河北大学《实验动物保护条例》,小鼠饲养在无特定病原体(SPF级)动物房中,期间自由进水进食,饲养环境温度为20~24 ℃.
1.2 方法
1.2.1 目的基因的获得
基于GenBank中hLeptin的基因序列(Accession No:NM_000230),选取其成熟肽基因序列作为克隆的对象,序列全长为438 bp,目的基因经密码子优化,并合成在pET-30a(+)质粒上,由金唯智生物科技有限公司完成.
1.2.2 原核表达载体的构建
根据hLeptin成熟肽基因片段序列,利用NEB Builder软件(https://nebuilder.neb.com/)设计相应引物(表1),下划线为线性化pET-30a(+)-SUMO载体的同源序列.使用hLeptin引物F和R扩增基因片段,PCR反应体系:2×Pfx mix 12.5 μL,模板0.5 μL,正反向引物(10 μmol/L)各0.5 μL,无酶ddH2O 11 μL.反应条件:98 ℃预变性3 min,98 ℃ 30 s,64 ℃ 30 s,72 ℃ 4 min,40个循环;72 ℃ 5 min.PCR产物经质量分数为0.8%的琼脂糖凝胶电泳检测后回收纯化目的基因片段.将hLeptin成熟肽基因片段与线性化pET-30a(+)-SUMO载体进行同源重组反应,反应体系:载体1 μL,目的片段4 μL,2×Clone mix 5 μL,充分混匀后,50 ℃反应1 h.将重组体转化至E.coliDH5α感受态细胞中,经BamH Ⅰ和Hind Ⅲ双酶切验证并筛选阳性单克隆后送至华大基因进行测序,得到重组质粒pET-30a(+)-SUMO-Lep.

表1 本研究中所使用的引物Tab.1 Primers used for clone in this study
1.2.3 细菌培养及目的蛋白最适温度的诱导表达
将构建正确的重组质粒pET-30a(+)-SUMO-Lep转化至表达E.coliBL21(DE3)感受态细胞中,涂布于含有硫酸卡那霉素(kanamycin sulfate,Kan)的 LB 平板上,培养箱中 37 ℃ 过夜培养.挑取单克隆于2 mL的LB液体培养基中(50 mg/mL Kan),37 ℃,220 r/min振荡培养.以体积分数为1%的接种量接入2 mL的LB液体培养基中(50 mg/mL Kan),分别在37、30、25、20 ℃ 培养至OD600为 0.6 时加入诱导剂IPTG,使其终浓度为 0.1 mmol/L,之后在各温度分别培养,220 r/min条件下培养 4、8、12、20 h.12 000 r/min离心2 min收集菌体,用1 mL的Lysis buffer(10 mmol/L NaH2PO4、300 mmol/L NaCl、50 mmol/L咪唑;pH 8)洗涤重悬菌体,再次离心,利用1 mL的Lysis buffer重悬菌体,进行超声波破碎.将破碎后的细菌裂解物在4 ℃下12 000 r/min离心 10 min,上清液即为含有可溶性表达蛋白的粗提液,同时将沉淀用1 mL的Lysis buffer重悬.通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamidegel electrophoresis,SDS-PAGE)检测各个温度下的表达效果.
1.2.4 hLeptin蛋白的纯化及浓度测定
将200 mL诱导完成后的菌液6 000 r/min离心10 min进行富集,取10 mL Ni-NTA Lysis buffer 重悬并收集菌体,6 000 r/min离心10 min,再用100 mL Ni-NTA Lysis buffer (50 mmol/L NaH2PO4、300 mmol/L NaCl、10 mmol/L 咪唑;pH 8) 将菌体重悬,用高压均质机进行破碎.破碎压力为0 MPa 3次,40 MPa 3次,80 MPa 3次,100 MPa 3次.将高压破碎后的细胞裂解液12 000 r/min、4 ℃离心 10 min,收集上清液粗提液.
使用镍柱亲和层析法对目的蛋白进行纯化,首先使用10个柱体积Ni-NTA的 Lysis buffer平衡镍柱,然后将上清液粗提液缓慢加入镍柱;再加入5个柱体积Ni-NTA 的Washing buffer (50 mmol/L NaH2PO4、300 mmol/L NaCl、50 mmol/L咪唑;pH 8)洗脱杂蛋白,最后用Ni-NTA 250 mmol/L Elution buffer(50 mmol/L NaH2PO4、300 mmol/L NaCl、250 mmol/L咪唑;pH 8)和Ni-NTA 350 mmol/L Elution buffer(50 mmol/L NaH2PO4、300 mmol/L NaCl、350 mmol/L咪唑;pH 8)对目的蛋白进行洗脱并收集,得到纯化后的产物,利用SDS-PAGE对目的蛋白的纯化效果进行检测.运用BCA法以BSA作为标准蛋白测定hLeptin相对蛋白浓度.
1.2.5 瘦素的生物活性鉴定
小鼠减肥实验:将12只健康雄性KM小鼠随机分为对照组和实验组(n=6).正常饲养7 d后,按每g体质量1 μL,实验组给与hLeptin (2.5 μg/μL),对照组给与PBS溶液(0.01 mol/L),每天上午9:00和下午5:00腹腔注射,持续1周.每日定时称体质量,并观察记录食物摄入量.比较2组小鼠体质量和摄食结果的差异.
成熟hLeptin蛋白对HepG2细胞的增殖作用:将生长状况良好的HepG2细胞用质量分数为0.25%的胰酶消化,并用含体积分数10%血清的DMEM培养液调整HepG2细胞浓度为5×104/mL后接种96孔细胞培养板 (每孔100 μL).贴壁生长后换无血清DMEM培养液,继续培养24 h,使细胞同步化.吸除细胞培养板孔中培养基,分别加入含hLeptin质量浓度为0、50、100、200、400 ng/mL的无血清DMEM培养基,每个质量浓度设6个平行孔,其中无hLeptin(0 ng/mL)为对照组.继续培养12 、24 h后,向待测孔各加入10 μL的CCK8,孵育1.5 h,用酶标仪测定450 nm波长处每孔的吸光度值A(以不加细胞的空白孔调零),计算细胞增殖率.
细胞增殖率=(A实验-A调零)/(A对照-A调零)×100%.
1.3 统计学方法

2 结果
2.1 表达质粒的构建
以带有合成基因的pET-30a(+)-Lep质粒和pTWIN1质粒为模板,分别扩增hLeptin基因和SUMO标签片段,结果如图1a所示,SUMO片段全长296 bp,带有hLeptin片段的质粒全长5 767 bp.利用Gibson Assembly无缝克隆,成功构建了重组载体pET-30a(+)-SUMO-Lep,用BamHⅠ和Hind Ⅲ进行酶切验证的结果如图1b所示,与预期结果一致.将得到的阳性克隆送至华大基因进行测序验证,测序结果经DNAMAN软件与pET-30a(+)-SUMO-Lep拼接图谱序列比对正确,表明质粒构建成功.
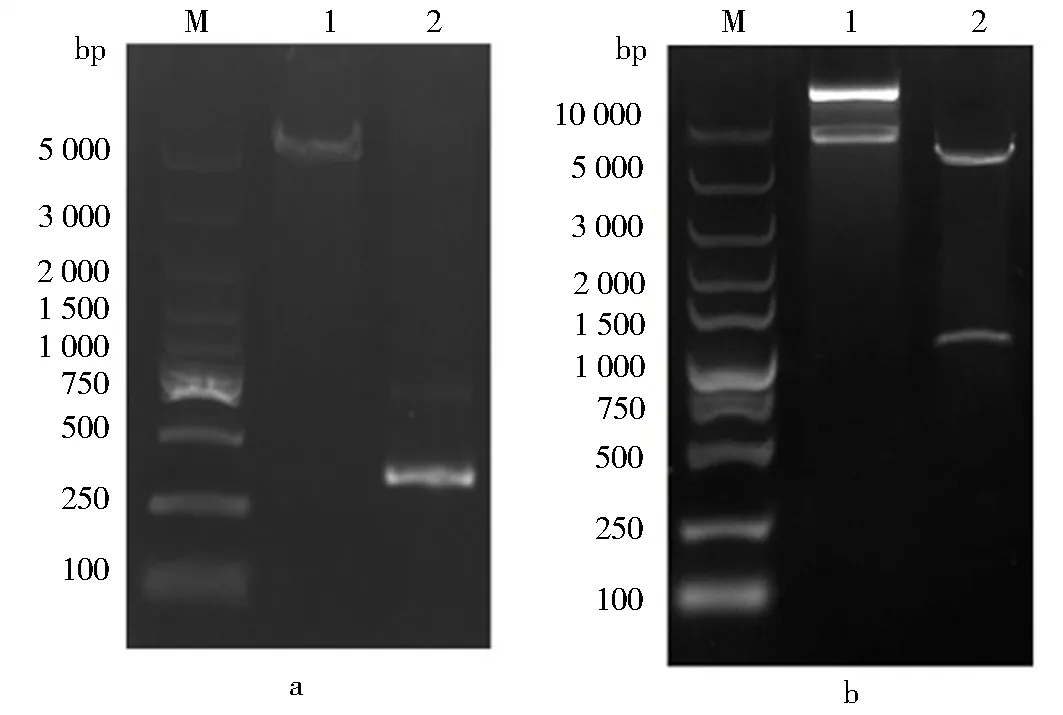
a.PCR扩增产物:M.Marker;1.hLeptin基因及载体;2.SUMO标签片段.b.双酶切验证无缝克隆产物:M.Marker;1.无酶切质粒;2.酶切后大片段为载体,小片段为插入序列.图1 重组表达质粒pET-30a(+)-SUMO-Lep的构建Fig.1 Construction of recombinant expression plasmid pET-30a(+)-SUMO-Lep
2.2 瘦素的可溶性表达及纯化
对含有重组质粒pET-30a(+)-SUMO-Lep的大肠杆菌在不同温度下诱导表达,结果如图2所示,在30 ℃条件下,上清液中带有SUMO标签的hLeptin表达量最高.

M.Maker;1.BL21(DE3) 上清液;2.BL21(DE3) 沉淀;3.37 ℃ 上清液;4.37 ℃ 沉淀;5.30 ℃ 上清液;6.30 ℃ 沉淀;7.25 ℃ 上清液;8.25 ℃ 沉淀;9.20 ℃ 上清液;10.20 ℃ 沉淀.图2 SDS-PAGE 分析不同诱导温度下 hLeptin 的表达水平Fig.2 SDS-PAGE analysis of the expression levels of hLeptin under different induction temperatures
通过Ni-NTA亲和柱纯化后结果如图3所示,成功对重组hLeptin进行了表达及纯化.纯化后的带有SUMO标签重组hLep的蛋白分子质量约为31.5 ku;利用SUMO特异性蛋白酶ULP去除SUMO标签的重组hLeptin蛋白分子质量为16 ku,与预期分子质量一致.
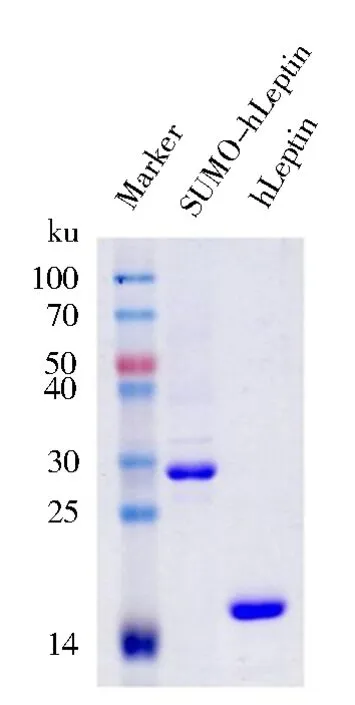
图3 纯化后的重组hLeptinFig.3 Purified recombinant hLeptin
2.3 重组hLeptin的质量浓度测定
利用BCA法测得562 nm波长下不同质量浓度标准蛋白溶液吸光度,根据表2得到标准曲线方程:y=0.475 8x+0.083 9,R2=0.991 9,线性关系良好(图4),稀释10倍后重组hLeptin样品的A562=0.289,将其代入标准曲线方程得到原hLeptin质量浓度为4.31 mg/mL.
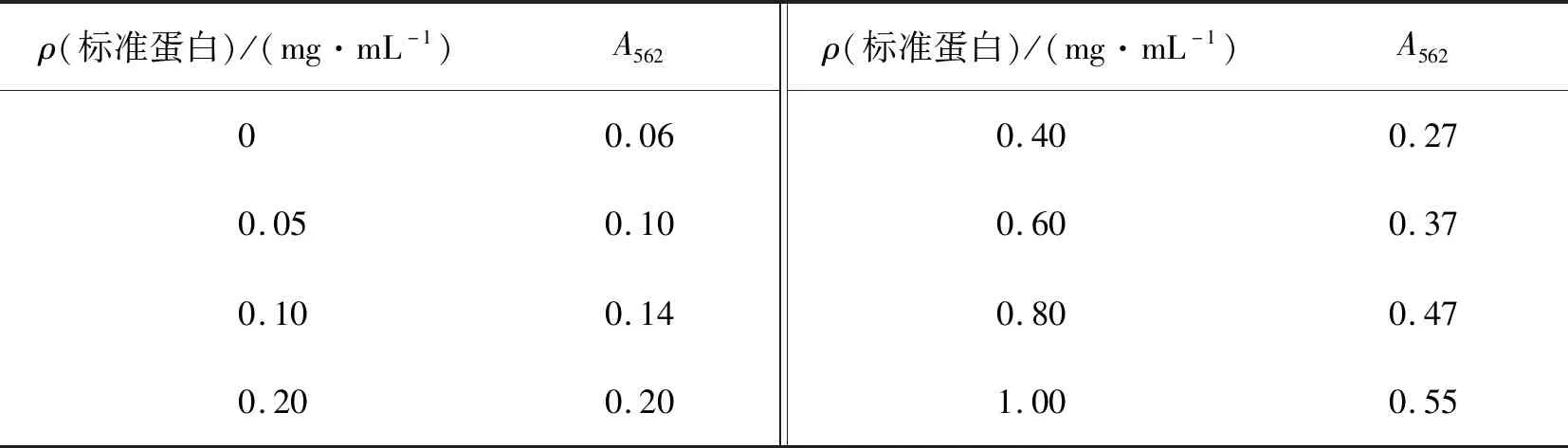
表2 不同质量浓度的标准品的吸光度值Tab.2 Absorbance values of the standard protein with different mass concentrations
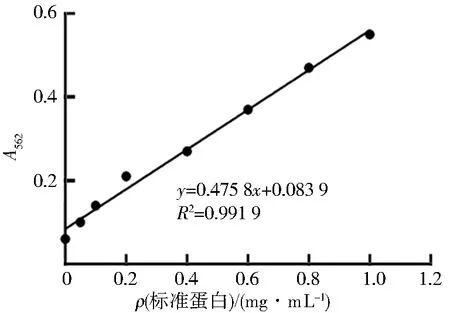
图4 标准曲线方程Fig.4 Standard curve equation
2.4 重组hLeptin的生物活性鉴定
小鼠腹腔注射hLeptin后第2天开始体质量增长量低于对照组,hLeptin组小鼠的进食量受到抑制,整个实验期内hLeptin组小鼠的体质量增长量和进食量均小于对照组(图5~6),差异具有统计学意义(P<0.05),由此可见重组表达的hLeptin具有良好的生物学活性.
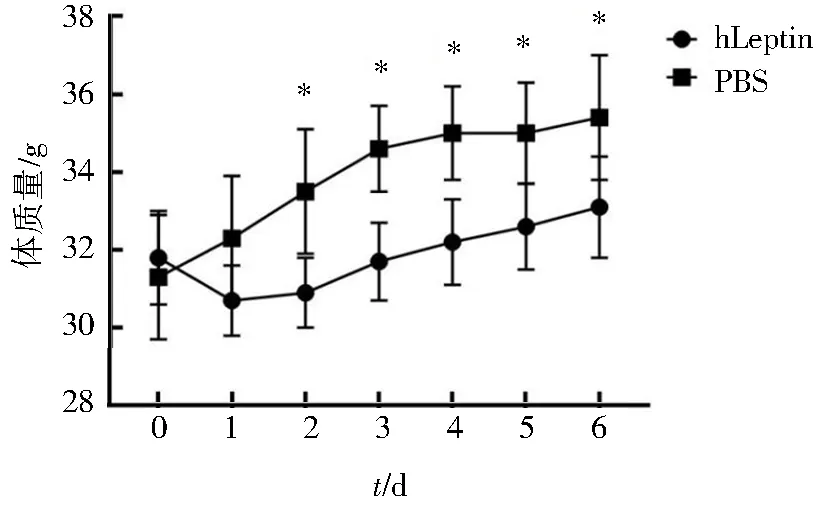
*P<0.05,hLeptin vs PBS图5 KM小鼠注射hLeptin后体质量的变化Fig.5 Changes of body mass after hLeptin injected of KM mouse

图6 KM小鼠注射hLeptin后摄食量的变化Fig.6 Changes of food intake after hLeptin injected of KM mouse
分别用0、50、100、200、400 ng/mL的hLeptin处理人肝癌HepG2细胞,12、24 h后用CCK8法检测细胞活力,与对照组(0 ng/mL)相比,hLeptin处理后的人肝癌HepG2细胞增殖水平均随着hLeptin质量浓度的增加而明显增加(图7),且差异具有统计学意义(P<0.05),表明瘦素可以诱导HepG2细胞增殖,证明重组表达的hLeptin具有良好的生物学活性.
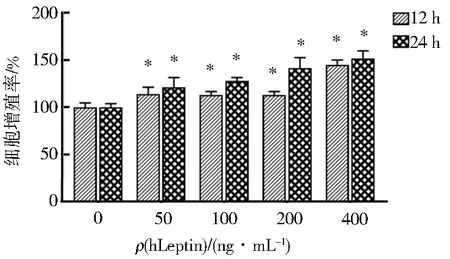
与对照组(0 ng/mL)相比*P<0.05.图7 不同质量浓度瘦素作用HepG2细胞不同时间对细胞增殖的影响Fig.7 Effects of different mass concentrations of hLeptin on cell proliferation of HepG2 at different time
3 讨论
DNA重组技术使体外表达目的蛋白成为可能[11].真核表达蛋白系统尽管具有帮助重组真核蛋白翻译后准确折叠和修饰的酶系统,能获得生物活性蛋白,但细胞培养周期长、蛋白得率低、操作要求高等缺点使其不能满足对生物活性蛋白的大量需求.以大肠杆菌为代表的原核表达系统具有遗传背景清晰、生长快速、表达量高、易于操作等优点,在科研、生物医药等领域已得到广泛应用.但真核蛋白在原核宿主细胞中的表达常会遇到外源蛋白错误折叠以无生物活性不可溶的包涵体形式存在的问题[12].如何通过提高外源蛋白表达的可溶性,获得具有生物活性的目的蛋白已成为原核表达系统广泛应用的最大瓶颈[13].
近年来SUMO常作为融合标签促进外源重组蛋白的表达,主要基于SUMO/SUMO特异性蛋白酶相互作用的机理[14].SUMO被发现可用作分子伴侣不仅能够提高蛋白在大肠杆菌的表达量,而且能够大大增加蛋白的可溶性,其作用机制可能是SUMO蛋白作为一个高度疏水的核心,为目的蛋白的折叠提供成核位点,促进蛋白间的相互作用并使其正确折叠,最终增强了融合蛋白的可溶性[15].此外,利用SUMO标签表达融合蛋白的另一个优点是利用SUMO特异性蛋白酶具有高度特异性,可以识别完整的SUMO 标签蛋白,并能高效地把SUMO从融合蛋白上切割下来,而且经过切割的目标蛋白不会携带多余的氨基酸残基,以产生具有所需N末端的天然蛋白[16].
人类与小鼠的Leptin同源性高达84%[17].在人类和啮齿动物中,血浆Leptin的水平反映了脂肪组织库的充盈状态,与体脂总量呈正相关[18].Leptin通过与包括中枢和外周的多个位点的瘦素受体特异性结合后,继而激活下游一系列信号转导通路而发挥其生理作用,因此Leptin具有广泛的生物学效应[19].大部分肥胖患者体内Leptin水平异常增高,表现为高瘦素血症及瘦素抵抗[20].Leptin参与了肥胖相关的肝癌的发生发展,多项研究指出[21-23],Leptin通过与人肝癌HepG2细胞上的瘦素受体结合促进肝癌细胞DNA合成和增强有丝分裂活性,从而诱导肝癌细胞增殖.而在临床中,由于Leptin基因突变而导致的先天性Leptin完全缺乏患者,在生命早期就会发展为极端肥胖,同时伴有明显的神经内分泌异常,通过补充重组hLeptin蛋白可改善患者病态肥胖及代谢异常[24].LEE的研究表明[25],重组hLeptin替代治疗可以改善高活性抗逆转录病毒疗法(HAART)所致的脂肪萎缩和代谢综合征患者的胰岛素抵抗和高脂血症.本研究成功建立了pET-30a(+)-SUMO原核表达系统,成功获得可溶性表达高活性的重组hLeptin.在对重组hLeptin进行生物学活性鉴定时,给予KM小鼠外源重组hLeptin后显著抑制了小鼠的体质量及进食量的增长;同时,观察到不同浓度重组hLeptin诱导人肝癌HepG2细胞增殖,与对照组相比差异显著.这些结果表明,本研究获得的重组hLeptin在体内和体外实验中均具有良好的生物学活性.
综上所述,本文利用SUMO融合标签对原核表达系统中促蛋白可溶性表达进行了研究,并对得到的重组hLeptin进行体内动物实验和体外细胞实验检测其生物学活性.成功表达了可溶且具有生物活性的hLeptin,为大规模生产hLeptin提供了一种新方法,这对于深入研究其在基础医学及临床医学中的作用具有重要意义,同时为广泛应用于表达可溶重组蛋白研究提供了一定的思路.
