Swyer综合征合并性腺母细胞瘤及无性细胞瘤临床病理分析
2022-10-19张雁瑞孔艳青史健梁冠男张梦琪
张雁瑞,孔艳青,史健,梁冠男,张梦琪
(南方医科大学附属深圳妇幼保健院病理科,深圳 518028)
Swyer综合征又称46,XY单纯性腺发育不全,是一种罕见的性别发育异常(DSD)疾病,由Swyer[1]于1955年首次报道,典型临床特征为:患者社会性别为女性,常以原发性闭经就诊;青春期第二性征发育不良,外生殖器为女性表型,发育幼稚,有输卵管、子宫及阴道,子宫发育不良,卵巢缺如,代之以条索状性腺;患者血清促性腺激素水平升高,性染色体核型为46,XY。条索状性腺发生肿瘤尤其是恶性生殖细胞肿瘤的风险高。由于该病发病率极低,临床及病理医生对其的认识更多来自病例报道,易导致对疾病认识不足,引起误诊、漏诊,从而错过治疗的最佳时机。本文选择2015年1月至2021年12月南方医科大学附属深圳妇幼保健院诊治的Swyer综合征患者3例(均合并性腺母细胞瘤及无性细胞瘤),回顾性整理、分析患者的临床表现、实验室检查、影像学检查、病理诊断及治疗方法等,并结合国内外相关文献进行讨论总结,探讨该病的诊断及治疗,以期减少误诊率,避免严重并发症。
一、病例资料
1.一般资料:3位患者社会性别均为女性,15~18岁,智力正常,身高及体重高于同龄人,均因青春期原发性闭经就医。无家族遗传史,母亲孕期无特殊用药史。
2.体格检查:外阴女性表型,发育幼稚,乳腺不发育或发育欠佳(表1)。
3.实验室检查:外周血染色体核型分析均为46,XY。血清卵泡刺激素(FSH)及黄体生成素(LH)水平均有升高,睾酮(T)及雌二醇(E2)水平降低。2例患者检测了抗苗勒管激素(AMH)水平并均显示水平降低。肿瘤标志物:糖类抗原(CA125、CA19-9、CA15-3)、甲胎蛋白(AFP)及癌胚抗原(CEA)正常。基因检测:病例3检测到SRY基因4个碱基缺失(表1)。
4.影像学检查:彩超及盆腔核磁均显示存在阴道、子宫及输卵管,子宫体积小,双侧卵巢细小或占位(表1)。
5.手术治疗:3例患者均行全麻下经脐单孔腹腔镜双侧输卵管及性腺切除术。术中见子宫幼稚,双侧性腺呈条索状(表1)。
二、病理特征
术中切除标本经10%中性福尔马林固定,石蜡包埋,常规制片,HE染色,光镜观察。免疫组化采用SP法(所用一抗包括CK、Vimentin、α-inhibin、Calretinin、CD99、SALL4、OCT4、PLAP、CD117、D2-40),DAB显色。每项均设有阳性及阴性对照,阳性对照以相应抗体的已知阳性切片作对照,阴性对照以PBS代替一抗作对照。所用抗体及试剂盒均购自福建迈新公司。
1.巨检:送检标本均为(左输卵管+左侧性腺)(右输卵管+右侧性腺)(表2、图1A)

表1 3例Swyer综合征患者临床特征

表2 双侧输卵管及性腺病理资料
2.HE染色:性腺母细胞瘤由被纤维结缔组织间质围绕的大小不等的细胞巢聚集而成,伴有明显钙化,钙化团块光滑,类圆形(图1B)。细胞巢实性,圆形或卵圆形,巢内见生殖细胞和类似未成熟Sertoli细胞或颗粒细胞混杂排列。生殖细胞大,胞浆丰富淡染,核大而圆,核仁明显;性索细胞小,核深染,卵圆形或胡萝卜形。性索细胞显示3种排列方式:(1)沿细胞巢外围呈花冠状排列;(2)围绕单个或团状生殖细胞排列;(3)围绕小圆形间隙排列,腔内含透明变性的嗜伊红无定型物,类似Call-Exner小体(图1C)。部分细胞巢周围见透明变性的基底膜样物质,细胞巢旁间质中散在多少不等的胞浆嗜伊红的Leydig细胞,单个或小簇状分布(图1D)。性腺母细胞瘤巢外间质中见条索状、岛状及巢片状排列的无性细胞瘤,瘤细胞形态与性腺母细胞瘤巢中的生殖细胞相同,可见核分裂像。肿瘤周围包饶含有淋巴细胞的纤维间质,淋巴细胞数量不等,偶尔见含生发中心的淋巴滤泡(图1E)。
3.免疫组化(IHC)染色:性腺母细胞瘤内性索细胞:Vimentin(+)、α-inhibin(+)、Calretinin(+)、CD99(+);性腺母细胞瘤内生殖细胞及无性细胞瘤细胞:CK(部分+)、SALL4(+)、OCT4(+)、PLAP(+)、CD117(+)、D2-40(+)(图1F、1G)。
4.病理诊断:病例1、2双侧性腺及病例3左侧性腺:性腺母细胞瘤合并无性细胞瘤;病例3右侧性腺:卵巢皮质样结构及脂肪纤维结缔组织(图1H)。所有输卵管均未见异常改变。
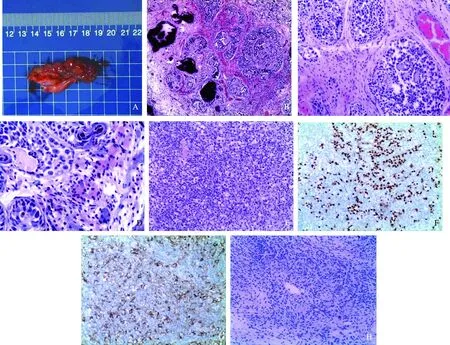
A:切除性腺,呈条索状外观;B:性腺母细胞瘤伴明显钙化(HE ×40);C:生殖细胞与性索细胞混杂排列(HE ×100);D:瘤巢周围间质中的Leydig细胞(HE ×200);E:无性细胞瘤(HE ×100);F:无性细胞瘤 OCT4(+)(IHC ×100);G:无性细胞瘤D2-40(+)(IHC ×100);H:病例3右侧性腺卵巢皮质样结构(HE ×100)。所有图片均出自本文病例。图1 切除性腺大体观及组织学检查
三、讨论
性别发育异常(disorder of sex development,DSD)是一种先天性染色体、性腺和表型性别的发育异常或不匹配[2],发生率大约1/5 500~1/4 500[3]。2006年欧洲和美国儿科内分泌协会达成共识[4],基于细胞遗传表型、激素水平、性腺组织学及临床特征,将DSD分为3组:46,XY DSD、46,XX DSD及性染色体DSD,其中46,XY DSD的发病机制最为复杂,又进一步分为2类,即性腺(睾丸)分化发育异常及雄激素合成、作用、代谢障碍[5-6]。自此,“假两性畸形”、“雌雄间性人”及“性反转”等术语不再使用。
Swyer综合征,又称为46,XY单纯性腺发育不全,属于性腺(睾丸)分化发育异常,因1955年Swyer[1]首次报道的2例“性反转”病例而得名。不同于当时被称为“男性假两性畸形”的临床表现,这两位“女性”因原发性闭经就医,她们身材高大,乳房轻微发育或不发育,有腋毛及阴毛,外生殖器为女性表型,有正常的阴道及小子宫,没有明显的附件,外周血染色体核型为(46,XY)。Swyer综合征发病率大约1/100 000[7],发病机制主要为性别分化过程中相关基因发生突变,导致性腺在胚胎不同时期发生不同程度的发育不全或退化,造成性别发育异常。目前已发现的相关基因有SRY、SF-1、SOX9、WT-1、MAP3K1、GATA4、FOG2、ATRX、DMRT1、DHH等[8-11],其中SRY基因作为性别决定基因,发挥着重要作用。性腺分化发育是一个由众多基因协同调控的非常复杂的过程。在胚胎发育第7周,由SRY基因编码的SRY蛋白促进睾丸的分化、发育,形成成熟的Sertoli细胞,一方面诱导Leydig细胞分化成熟并产生睾酮,促进睾丸曲细精管、输精管及附睾的发育,另一方面合成抗苗勒管激素(AMH),导致苗勒管退化。若SRY基因缺失或异常表达则导致睾丸发育障碍,苗勒管得不到抑制而进一步分化为输卵管、子宫及阴道上1/3段。本组病例中有2例做了性分化相关基因检测,其中1例检测到SRY基因4个碱基缺失,另外1例未检测到明确致病突变。
本组3例患者均有Swyer综合征的典型临床特征:社会性别为女性,智力正常,身材高大,以青春期原发性闭经就诊;第二性征发育不良,外生殖器为女性表型,发育幼稚,有输卵管、子宫及阴道,子宫发育不良,卵巢缺如,代之以条索状性腺;患者血清卵泡刺激素及黄体生成素水平升高,睾酮及雌二醇水平降低;外周血染色体核型检查为46,XY。条索状性腺发生肿瘤的风险高,最常见的肿瘤类型为一种良性肿瘤——性腺母细胞瘤,发生率约为30%[12],单侧或双侧发生,往往在青春期疾病得到诊断时已伴发,有报道在婴幼儿时期可发生[10]。本病的条索状性腺恶变率高,是性别发育异常疾病中最易恶变的病种,原因可能为发育不全的性腺组织与腹腔内环境相互诱导促发所致。恶变肿瘤为恶性生殖细胞肿瘤,如无性细胞瘤、未成熟型畸胎瘤、胚胎性癌及卵黄囊瘤。50%~60%的性腺母细胞瘤与恶性生殖细胞肿瘤相关,大多表现为无性细胞瘤[13]。本组3例患者性腺母细胞瘤均伴发无性细胞瘤,从相当程度上说明无性细胞瘤在性腺恶性肿瘤中发生率高。性腺肿瘤的发生风险会随年龄的增长而增加,因此,早期发现、早期诊断并行预防性双侧性腺切除以降低肿瘤发生风险尤为重要。
性腺母细胞瘤罕见,几乎全部发生于性腺发育不全患者,偶尔见于其他方面皆正常的卵巢。大多数病理医师,尤其是基层医院的病理医师对该肿瘤的认识仅限于参考书。性腺母细胞瘤为实性肿瘤,大小不一,从显微镜下病变到数厘米不等,颜色从黄、灰黄到棕色不等。组织学形态独特,在纤维结缔组织间质中见到大小不等的细胞巢聚集,常伴明显钙化或几乎完全钙化。钙化是此肿瘤的典型特征,钙化团块光滑,类圆形。细胞巢实性,圆形或卵圆形,偶尔可以较大,并拉长。巢内见生殖细胞和类似未成熟Sertoli细胞或颗粒细胞混杂排列。生殖细胞大,胞浆丰富淡染,核大而圆,核仁明显,可见核分裂像;性索细胞小,核染色深,形状为卵圆形或轻度拉长的胡萝卜样。性索细胞显示3种排列方式:沿细胞巢外围呈花冠状排列;围绕单个或团状生殖细胞排列;围绕小圆形间隙排列,腔内含透明变性的嗜伊红无定型物,类似Call-Exner小体。细胞巢周围常见到透明变性的基底膜样物质,这是性腺母细胞瘤的另一典型特征。约66%的病例于细胞巢旁间质中见到多少不等的Leydig细胞或黄素化细胞,单个或小簇状散在分布。在伴发无性细胞瘤的病例中,性腺母细胞瘤巢外间质中见到条索状、岛状及巢片状排列的无性细胞瘤,瘤细胞形态与性腺母细胞瘤巢中的生殖细胞相同,可见核分裂像。肿瘤被数量不等的淋巴细胞分隔,偶尔见到含生发中心的淋巴滤泡。性腺母细胞瘤因其独特的组织学形态,不易与其他性腺肿瘤相混淆,在病理诊断中唯一需要注意鉴别的是混合性生殖细胞-性索间质细胞肿瘤,后者肿瘤形态不一,无细胞巢团结构,无钙化及透明变性,鉴别并不困难[14]。
Swyer综合征是一种罕见疾病,其诊治涉及妇科肿瘤、内分泌及心理等多专业,需多学科合作综合治疗,包括性别的选择、切除发育不全的性腺组织、性激素替代治疗以及多学科团队长期随访[15]。患者虽染色体性别为男性,但生殖器表型为女性,社会性别及心理性别也为女性,因此患者意愿多为继续维持女性性别。因青春期前发现此病的几率很低,有研究建议14岁女性如果第二性征不发育或仍未月经来潮,需引起重视,尽早就医进行体格、影像、性激素水平及染色体核型检查[10]。因本病的条索状性腺恶变率高,一旦确诊需尽早切除发育不全的性腺组织,并保留子宫以备日后辅助生殖。术后给予性激素替代治疗,雌、孕激素长期序贯疗法可诱导青春期第二性征发育,促使患者月经来潮,使原本发育不良的子宫进一步生长发育,为后期辅助生殖打好基础[16]。本病一旦确诊,医师还需考虑患者的心理状态,注意与其沟通的方式和技巧,充分考量患者在生理上和心理上所面临的问题,进行适当的心理疏导,与患者及家属充分沟通,共同制订治疗方案,并做好术后长期随访工作。本组3例患者均选择继续维持女性性别,并切除发育不全的双侧性腺,术后给予性激素替代治疗,并进行长期随访。
