一种高效的β 地中海贫血CD17(A>T)点突变293T 细胞系的建立
2022-10-16刘永祥蔡炳许言曾艳红周少虎麦庆云
刘永祥,蔡炳,许言,曾艳红,周少虎,麦庆云*
1.广州中医药大学第一附属医院生殖医学科,广州 510405;2.中山大学附属第一医院生殖医学中心,广州 510080
β-地中海贫血是由于基因点突变或移码导致β珠蛋白肽链合成不足的一种遗传性血红蛋白病。据HbVar 数据库统计,全球已发现300 多种突变类型[1],其中,CD41/42(-CTTT)、CD17(A>T)和IVS2-654(C>T)构成的突变,是最常见的β 突变类型[2]。β 珠蛋白基因的CD17 (A>T)点突变是最早报道的与β 地贫相关的点突变之一。地中海贫血可通过骨髓或造血干细胞治疗,但费用昂贵且很难寻找匹配的供者。近年来随着CRISPR-Cas9 技术的发展,基因治疗已成为重要的方向,但编辑效率很低,实验重复性较差。“CORRECT”编辑技术的发现,在CRISPR/Cas9 靶向所需序列内插入同义突变碱基到同源定向修复(homology-directed repair,HDR)修复模板中,可以在很大程度上阻止不良的再次编辑,提高ssODNs 的整合效率[3,4]。因此,本研究拟利用“CORRECT”新技术获得β-地贫CD17(A>T)突变基因型的HEK293T 细胞系,以期建立一种高效的基因点突变新方法,为后续的基因功能研究以及基因修复方法提供更有力的技术支撑。
1 材料与方法
1.1 材料
1.1.1 细胞系和质粒 HEK293T 细胞购自ATCC 细胞 库,CRISPR/Cas9质粒PX459(pSpCas9-2A-Puro(PX459)V2.0)购自Addgene。
1.1.2 主要试剂BbsI、T7E I、KspA I、T4 连接酶、T4磷酸化酶(New England Biolabs);sgRNA 寡核苷酸链(生工生物);4D-Nucleofector™电转试剂盒(Lonza);单链供体寡核苷酸(Genewiz);DMEM 培养基(Gibco);FBS 胎牛血清(Hyclone)。
1.2 方法
1.2.1 sgRNA 的设计与Cas9-sgRNA 载体构建 (1)根据NCBI 数据库人类HBB(Gene ID: 3043)的基因序列,使用靶点在线设计网站https://zlab.bio/guidedesign-resources 设计sgRNA 靶点:HBB-sgRNA1(5’-CACGTTCACCTTGCCCCACA-3’)和HBB-sgRNA2(5’-CCTGTGGGGCAAGGTGAACG-3’)。在实验中,我们也把sgRNA1称为Correct1,sgRNA2称为Correct2。(2) 根据靶点序列合成正负链sgRNA 引物,并在引物5'端加上BbsI酶切位点的黏性末端,正向引物:CACC-(N)20,反向引物:AAAC-(N)20R。利用T4磷酸化酶进行5'端磷酸化修饰,并经退火(37 ℃,30 min;95 ℃,10 min)形成双链片段,用于后续实验。(3) 取100 ng经BbsI酶切线性化的PX459 载体与2 μL上述磷酸化后的sgRNA 双链用1 μL T4 连接酶,16℃过夜连接。再将连接产物转化入大肠杆菌DH5α 中,涂布于含氨苄抗性的LB 固体培养基37 ℃进行培养16~18 h。次日挑取单克隆菌落,37 ℃摇床扩增培养,提取质粒,测序鉴定阳性克隆。
1.2.2 ssODN 序列合成 ssODN 作为外源性同源重组修复模板,主要取sgRNA 靶点上下游各50 bp 序列,替换上CD17(A>T)点突变碱基,同时依据氨基酸的简并性,在PAM 序列附近引入同义突变碱基(G>T)。2 条ssODNs 如下(突变碱基用小写字母表示):正 向 donor1(5’-ATGGTGCATCTGACTCCTGAG GAGAAGTCTGCCGTTACTGCCCTGTGGGGCtAGGT tAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGC AGGTTGGTAT-3’)和反向donor2(5’-ATACCAA CCTGCCCAGGGCCTCACCACCAACTTCATCCACG TTaACCTaGCCCCACAGGGCAGTAACGGCAGACTT CTCCTCAGGAGTCAGATGCACCAT-3’)。
1.2.3 电转染 通过消化处理得到的单细胞悬液按照Lonza 转染试剂盒说明书,将轻轻混匀的电转混合液(82 μL 电转液I、18 μL 的电转液Ⅱ、PX459-sgRNACas9 共表达载体、donor)转移到电转杯中,上机程序参照说明书。电转后,将细胞悬液转移至无血清培养基中进行培养。
1.2.4 sgRNA 靶点有效性检测 为了验证不同靶点的切割效率和ssODN 的整合效率,将实验分为5组:①Correct1+donor1;②Correct1+donor2;③Correct2+donor1;④ Correct2+donor2;⑤以及阳性对照组Sacas9+L+R+saDonor。将各组电转染24 h 后用含有0.6 μg/mL Puro 的无血清培养基进行药筛,培养48 h后收取细胞,提取DNA 进行PCR 扩增出包含靶点序列在内的DNA 片段,最后取200 ng PCR 产物通过T7EI和KspA I双酶切实验,比较各组切割效率与整合效率。PCR 引物为HBB-FP:5'-GCAATTTGTACTGA TGGTATGG-3';HBB-RP:5'-ATAACAGCATCAGGAG TGGAC-3'。
1.2.5 单克隆细胞筛选与测序 根据酶切实验的结果,选择整合效率最高的实验组采用计数稀释接种的方法进行单克隆筛选,吸取部分扩大培养后的单克隆集落细胞按照DNA 提取试剂盒提取基因组DNA,PCR 扩增后测序鉴定。
2 结果
2.1 PX459-sgRNA-Cas9 共表达载体的构建
CRISPR/Cas9 原质粒PX459 大小为9175 bp,如图1 琼脂糖凝胶电泳条带2 所示,利用BbsI酶将PX459切成9153 bp 和22 bp 两段。条带1 原质粒有2 条条带,与质粒的超螺旋、开环和线性3 种构型吻合。条带2 酶切后质粒呈线性化状态,且片段大小符合预期位置。回收线性化的PX459 载体并分别与退火后的sgRNA 连接,转化DH5α,挑取单克隆、提取重组质粒并测序验证。
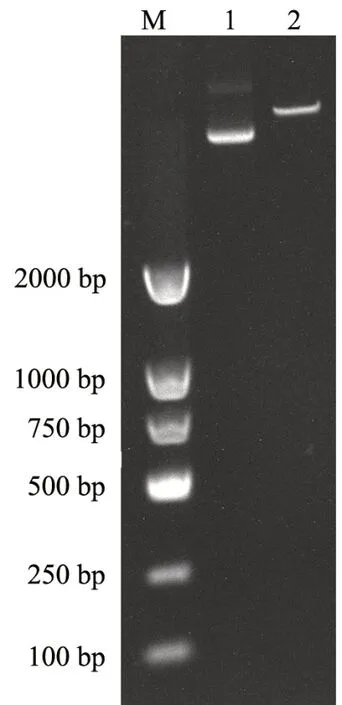
图1 PX459 质粒Bbs I酶切结果Fig.1 PX459 plasmid digestion by Bbs I enzyme
2.2 电转染效率
在HEK293T 细胞中电转染绿色荧光蛋白(Green fluorescent protein,GFP)质粒,24 h 后观察GFP 的表达情况,评估电转染效率(图2)。荧光显微镜下观察,电转染1 µg GFP 质粒细胞中GFP 表达最少,不到20%。而电转染2 µg 质粒则表达GFP 的细胞比例达到90%,转染效率较高。电转染3 µg 质粒表达GFP 的细胞比例达到95%以上。因此,为了减少电转染的毒性又达到较好的电转染效果,最终选择了电转染2 µg质粒的量进行后续实验。
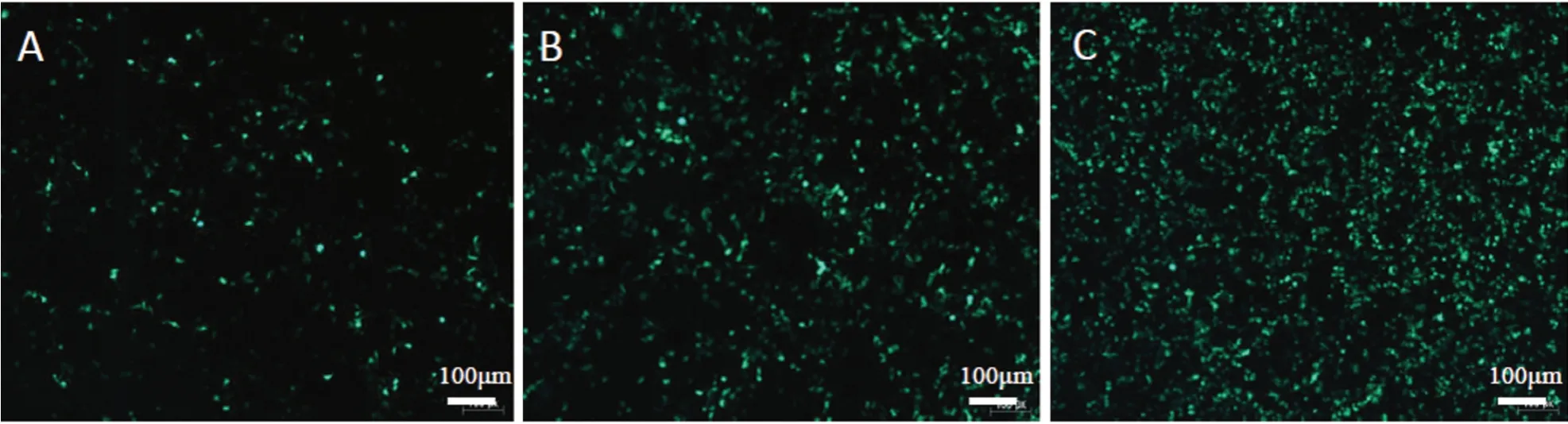
图2 HEK293T 细胞转染GFP 质粒的效率检测 A: 电转染1 µg GFP 质粒B: 电转染2 µg GFP 质粒C: 电转染3 µg GFP 质粒Fig.2 Transfected efficiency detection of GFP in HEK293T cells A: 1 µg GFP plasmid; B: 2 µg GFP plasmid; C: 3 µg GFP plasmid
2.3 HBB 基因CD17(A>T)点突变的HEK293T 细胞系的建立
2.3.1 双酶切鉴定 T7EⅠ酶切割效率实验:结果图3-A 中显示,PCR 总长度是578 bp,阳性对照组和各实验组均被T7EⅠ切割,实验组①~④酶切后分为3 条条带:原有条带578 bp,以及被切割后的340 bp 和238 bp 的两条带,结合灰度分析图3-C,共同说明4组实验组均有效率。
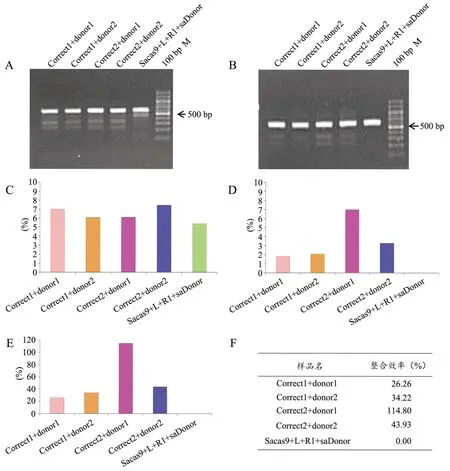
图3 不同靶点和donors 编辑效率和整合效率比较A: T7E I 酶切电泳图谱 B: KspA Ⅰ酶切电泳图谱 C: T7E I 酶切电泳条带灰度值分析 D: KspA Ⅰ酶切电泳条带灰度值分析E: 不同靶点和不同donor 的整合效率比较F: 各组整合效率汇总表Fig.3 Comparison of editing efficiency and integration efficiency of different targets and donorsA: Electrophoresis patterns after T7E I digestion; B: Electrophoresis patterns after KspA Ⅰdigestion; C: Gray value analysis of image strip after T7E I digestion; D: Gray value analysis of image strip after KspA Ⅰdigestion; E:Comparison of integration efficiency of different targets and donors; F: Table of integration efficiency for each group
Donor 的整合效率(KspA Ⅰ酶切)实验:当donor 整合至目标位置时,会被KspA Ⅰ酶识别并切割,如图3-B的电泳图所示,并结合图3-D 胶图灰度分析,对照组Sacas9组因donor 未引入KspA Ⅰ酶切位点,在切胶图上表现为未切割,作为对照证明了Correct 方案可行,排除了实验操作因素。结果显示,4组实验组均有KspA Ⅰ酶的切割效率,说明这4组均有donor 的整合,并图3-E 和3-F 所示,实验组③(Correct2+donor1)的整合效率最高,达到114.80%,而实验组①、②、④分别为26.26%、34.22%和43.93%。值得注意的是实验组③的整合效率计算后超过100%(理论上小于100%,因为切割后才有整合),可能是因为imageJ 软件自身的敏感度缺陷所致。计算公式:整合效率=(KspAⅠ酶切效率/T7EⅠ酶切效率)×100%,
2.3.2 测序鉴定 将野生型293T 细胞和四组实验组的PCR 产物进行Sanger 测序,如图4 所示,donor 作为同源模板与基因组DNA 发生了整合,这四组实验组均引入了β-地中海贫血基因型CD17(A>T)和同义突变碱基(G>T),因未行单克隆挑选,故显示为杂合的峰图。
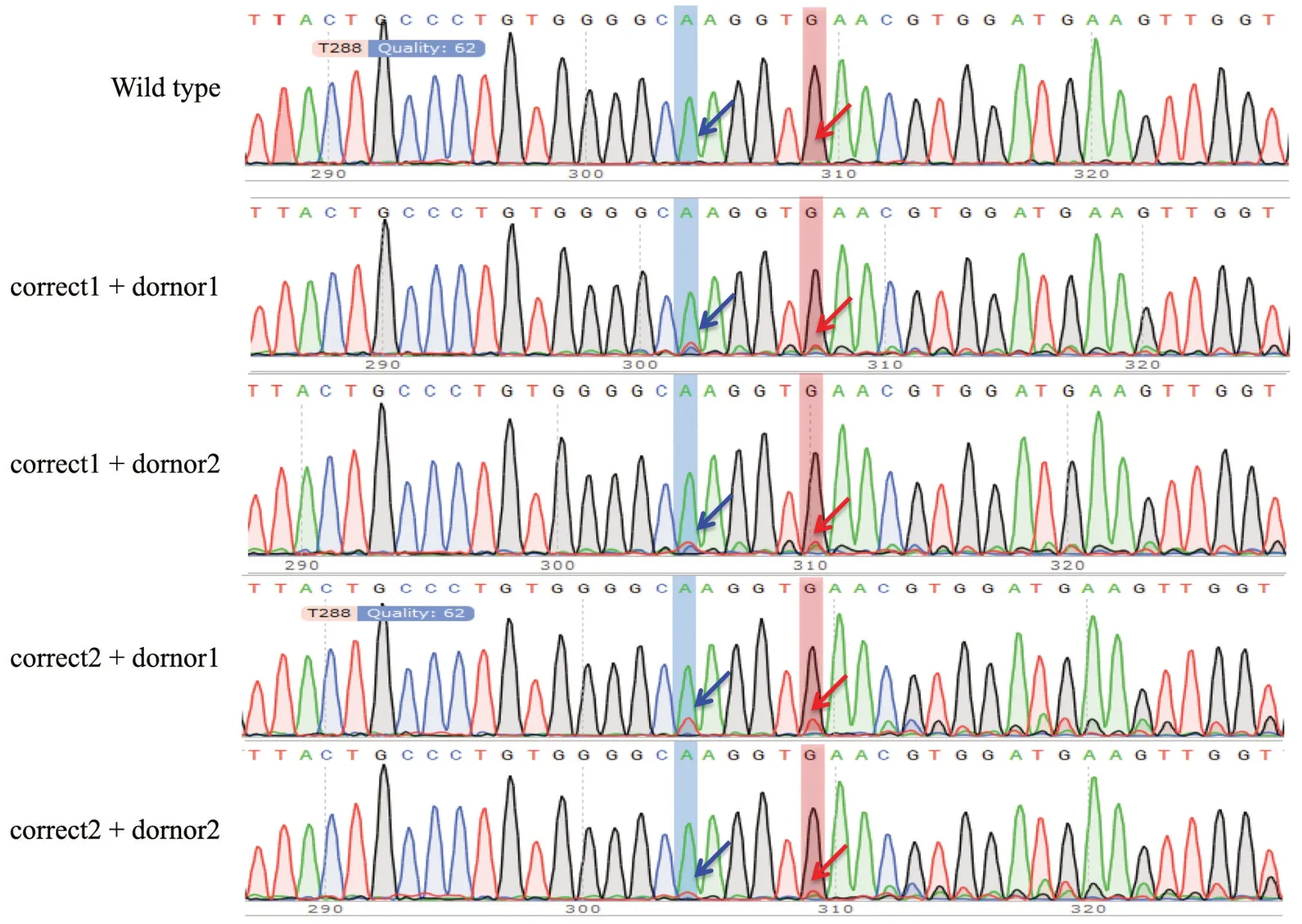
图4 各组总细胞提DNA 后进行PCR 测序的峰图蓝箭头示β17 致病点突变碱基(A>T) 红箭头示同义突变碱基(G>T)Fig.4 PCR sequencing after DNA extraction from total cells of each groupThe blue arrow showed β17 pathogenic point mutation base (A>T),and the red arrow showed synonymous mutation base (G>T)
2.4 地中海贫血β17(A>T)点突变的单克隆293T 细胞系的建立
根据酶切实验结果,实验组③的整合效率最高,故对实验组③的总体细胞进行单克隆筛选,PCR 扩增并测序鉴定。一共挑选12 个单克隆集落进行测序,获得1 株含纯合突变的单克隆细胞系,测序结果图5显示,单克隆293T 细胞系引入了β-地中海贫血CD17(A>T)和同义突变(G>T)碱基。另外,12 个单克隆中还包含2 个杂合突变以及9 个无相应碱基突变的野生型单克隆。
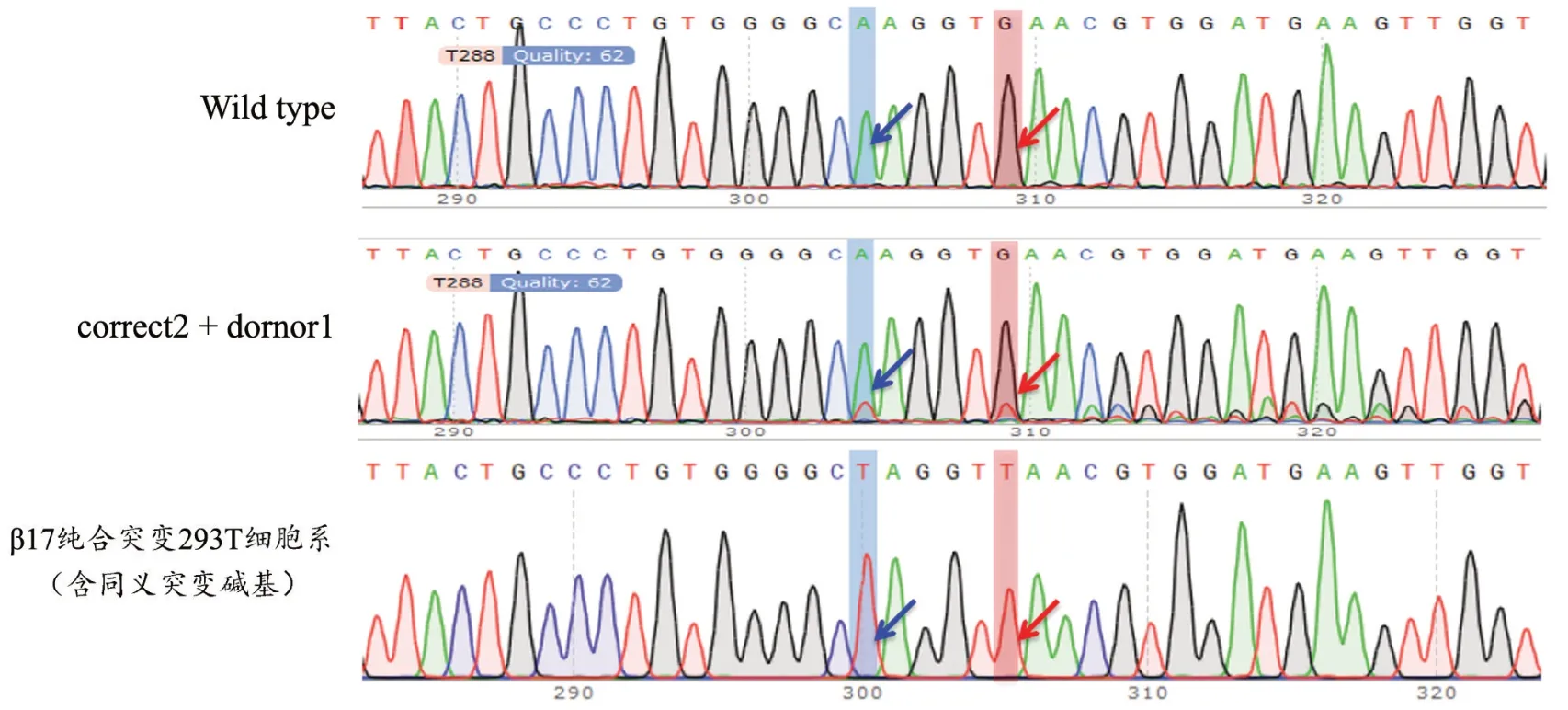
图5 野生型293T 细胞与β17 突变型单克隆293T 细胞系的测序峰图蓝箭头示β17 致病点突变碱基(A>T) 红箭头示同义突变碱基(G>T)Fig.5 Sequencing of wild-type 293T cells verse β17 mutant monoclonal 293T cellsThe blue arrow showed β17 pathogenic point mutation base (A>T),and the red arrow showed synonymous mutation base (G>T)
3 讨论
全球有超过50000 种人类遗传疾病,大部分是由基因点突变引起的。因此,对基因组点突变的研究不仅能揭示重要的生物学机理,更有利于人类疾病治疗手段的开发。CRISPR/Cas9 是一种新兴的基因编辑工具,与传统ZFN 和TALEN 两种人工核酸酶相比具有编辑效率高、操作简便、定点编辑等优点,因此也被广泛用于基因缺陷细胞和动物模型的构建。
构建点突变主要分为两类,一类是对基因组DNA 产生双链断裂并通过同源重组的方式引入突变位点。在sgRNA 的靶向定位下,DNA 双链被Cas9 精确剪切,如果引入一个携带靶片段同源臂和突变位点序列的模板,将会通过同源重组实现片段插入或定点突变[5]。第二类就是碱基编辑器,也属于CRISPR 技术的一种,通过对Cas 蛋白的改造,使其只具有切割DNA 单链的能力,结合碱基脱氨酶的催化活性,实现核酸链上特定位点碱基的改变[6]。但碱基编辑器的发展与应用尚不成熟,编辑效率较低,存在较高的脱靶率[7]。而且编辑窗口有限,无法进行任意四种碱基的互相替换,也无法解决碱基缺失和插入的基因致病突变类型,因此该方法仍需进一步的优化与研究。
目前报道的基因修复方法大多是以dsDNA 质粒为供体模板进行同源修复,但也有报道质粒供体部分骨架(在同源臂之外)也整合到目标基因组位点上[8]。这些不希望的骨架整合频率在不同的位点之间有很大的差异,据报道甚至“从0 到100%不等”[9]。与双链DNA(如质粒或病毒载体) 修复机制不同,以单链寡核苷酸(ssODN)作为供体模板,ssODN 不参与NHEJ,因此不太可能插入非特异性裂解位点,不太可能随机整合到基因组中,从而产生安全、精确的基因编辑[10]。Zorin等[11]发现,与质粒等dsDNA 供体模板相比,单细胞绿藻莱茵衣藻中的ssODN 模板随机整合到基因组中的倾向大大降低。此外,单链寡核苷酸更容易批量生产、更经济,具有高纯度和安全特性。因此,相对于利用dsDNA 供体或其他遗传操作进行小片段插入、缺失或点突变,利用ssODN 供体的CRISPR 技术是一种高效和引人注目的技术。在我们的实验中,则应用了“CORRECT”方法,在修复模板(ssODN)引入了同义突变的碱基,有效减少了Cas 蛋白的再次编辑,提高了编辑和整合效率。在我们的实验中,4组实验组的编辑效率均达到60%以上(图3-C)。而从整合效率来看,Correct2 靶点显示了较高的整合效率,达到40%以上。
利用CRISPR/Cas9 和ssODN 对人类遗传疾病基因进行组合编辑是修复和产生点突变的有效策略。在可靠地将DNA 改变引入已建立的细胞系之后,这项技术又被推广到体内外的研究。Aarts等[12]在小鼠胚胎干细胞(mouse embryonic stem,ES)细胞中,利用ssODNs 成功地在视网膜母细胞瘤(retinoblastoma,Rb)中实现密码子替换(N570F)。Niu等[13]指出,ssODN 与CRISPR/Cas9 结合能够纠正在β-地贫诱导型多能干细胞(iPSCs)中的HBB基因CD41/42 突变。而“CORRECT”是Tessier等[3,4]发现的新方法,他们在人类干细胞中无瘢痕地引入与疾病相关的纯合和杂合突变,并发现在编辑人iPSCs 时,高达95%的已编辑位点的序列会被再次编辑,造成额外的indel 突变破坏。因此,他们通过在PAM 序列(protospacer adjacent motif,PAM)附近整合能够有效地阻断多个位点的再次编辑的同义突变或致病突变,这些Casblocking 突变能以高效率与精度选择性引入单等位或双等位基因序列,使HDR 的编辑精度提高到每个等位基因的10 倍。在极端情况下,CORRECT 只需要挑选几百个克隆,而如果不采用阻断突变策略,则可能需要几万个克隆。他们还表示,最接近切割位点,其HDR 的整合效率最高。对于ssODN 的长度是否影响整合效率的问题,实验中我们与其他研究者类似[4,14],都使用短片段来进行单点突变的引入。在我们实验中应用了一段100 nt 的单链寡核苷酸(位于sgRNA 靶点上下游各50 nt),对于较高效率的Correct2(sgRNA2)靶点,实验显示正向的ssODN 整合效率更高。此外,有文献报道,对于短片段(100 bp 内)的插入,单链寡核苷酸要比双链具有更高的插入效率[15]。
对于CRISPR/Cas9 而言,CORRECT编辑后细 胞系的脱靶分析同样重要。但是,应用了同样的导向RNA,因此很可能不会扩大潜在脱靶位点的数量。但在多种细胞类型中多个基因组位点上的脱靶程度还有待阐明。本研究虽然没有Western blot 检测β 珠蛋白的表达,同时也没有进行了基因点突变后细胞的功能验证,但在方法学上为我们提供了一种高效的点突变的编辑方法,为后续的研究提供了坚实的模型基础。
本研究成功将“CORRECT”方法应用于293T 细胞系,获得高效的地中海贫血症β17 单碱基突变细胞系,发现同义突变的引入使得整合效率得到了极大的提高,并且有效减少在每个基因编辑阶段手工挑选单克隆的时间。本研究对单碱基突变细胞系以及单碱基突变动物模型的建立具有重要意义。
