高糖培养对大鼠肾小管上皮细胞中SIRT1和PGC-1α水平及线粒体功能的影响*
2022-10-14刘兴梅金红君沈燕何晓兰田禾
刘兴梅, 金红君, 沈燕, 何晓兰, 田禾
(1.贵州省人民医院 检验科, 贵州 贵阳 550002; 2.贵州省人民医院 肾内科, 贵州 贵阳 550002; 3.贵州省人民医院 病理科, 贵州 贵阳 550002; 4.贵州省临床检验中心 检验检测科, 贵州 贵阳 550002)
糖尿病肾脏病(diabetic kidney disease, DKD)伴有肾小球硬化、系膜细胞扩张以及肾间质纤维化等一系列病理变化,如不及时治疗,随着病程进展将发展为终末期肾病。研究发现,氧化应激与DKD密切相关,但其发病机制尚未明了[1]。过氧化物酶体增殖物激活受体γ共激活因子1α(peroxisome proliferator-activated receptor gamma coactivator 1-α,PGC-1α)是一种转录共激活因子,亦是线粒体生物发生和功能的主要调节因子。PGC-1α在高能量需求的组织中高表达,与糖脂代谢相关[2]。足细胞构成肾小球滤过屏障的外层,其能量需求依赖于与线粒体动力学密切相关的有效氧化呼吸。高糖可降低PGC-1α的表达,并降低足细胞中的线粒体DNA含量,从而改变线粒体生物发生和导致线粒体DNA损伤[3]。沉默配对型信息调节因子2同源物1(sirtuin 1,SIRT1)是一种多能分子,在细胞存活和凋亡、炎症应激以及细胞生长、分化、代谢等方面发挥着重要作用,通过改变底物的乙酰化状态来调节其转录活性和蛋白质表达水平,参与ATP的生成,从而改善线粒体功能障碍[4]。目前对肾小管上皮细胞,PGC-1α与SIRT1是否能通过保护线粒体从而减少高糖所致的细胞凋亡和细胞能量改变的研究甚少,因此,本研究通过体外培养NRK52E细胞,探讨在高糖环境下SIRT1和PGC-1α对NRK52E细胞中线粒体损伤及凋亡的作用及机制。
1 材料与方法
1.1 材料及试剂
NRK52E细胞购买于美国模式培养物集存库(American type culture collection, ATCC),抗体甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase, GAPDH)、PGC-1α、SIRT1、cleaved-Caspase-3(proteintech)、 腺嘌呤核苷三磷(adenosine-triphosphate, ATP)含量检测试剂盒(Solarbio)、线粒体膜电位检测试剂盒、活性氧(reactive oxygen species,ROS)检测试剂盒及超敏增强化学发光剂(enhanced chemiluminescence, ECL)化学发光试剂盒为碧云天产品。
1.2 研究方法
1.2.1细胞培养 NRK52E细胞株复苏:液氮罐中取出细胞,迅速放入37 ℃水浴箱融化,将细胞悬液加入含DMEM培养基的离心管,反复吹打,800 r/min离心5 min,小心吸弃上清,加含10% FBS 低糖DMEM培养基4 mL,重悬细胞,接种于25 cm2培养瓶,于37 ℃ 5% CO2培养箱培养。
1.2.2细胞分组及处理 将NRK52E细胞分为正常糖组(NG组,含糖5.5 mmol/L、10% FBS的DMEM培养)和高糖组(HG组,含糖30 mmol/L、10% FBS的DMEM培养),培养48 h后收集细胞。
1.2.3流式细胞仪检测线粒体ROS水平 按照1 ∶1 000用无血清培养液稀释活性氧荧光探针(DCFH-DA),使终浓度为10 μmol/L;细胞收集后悬浮于稀释好的 DCFH-DA中,细胞浓度为1.0×108~2.0×1010个/L,37 ℃细胞培养箱内孵育20 min。每隔3~5 min颠倒混匀一下,使探针和细胞充分接触。用无血清细胞培养液洗涤细胞3次,以充分去除未进入细胞内的DCFH-DA。
1.2.4流式细胞仪检测线 粒体膜电位(mitochondrial membrane potential,ΔΨm) 收集细胞用PBS洗涤细胞3次,收集不多于1×106个细胞,取10×Incubation Buffer 100 μL加灭菌去离子水900 μL稀释成1×Incubation Buffer,混匀并预热至37 ℃,吸取1×Incubation Buffer 500 μL,加入JC-1 1 μL旋涡混匀配成JC-1工作液;取JC-1工作液500 μL将细胞均匀悬浮,37 ℃、5% CO2的培养箱中孵育15~20 min,室温离心(2 000 r/min, 5 min)收集细胞,用1×Incubation Buffer 洗2次;吸取10×Incubation Buffer 500 μL重新悬浮细胞,流式细胞仪检测分析。
1.2.5紫外分光光度法检测线粒体ATP 先收集细胞到离心管内、弃上清,比例为细胞数量(104个) ∶提取液体积(mL)=(500~1 000) ∶1(建议500万细胞加入提取液1 mL), 超声波破碎1 min(冰浴,强度 20%或200 W,超声2 s,停1 s),12 000 r/min 4 ℃离心10 min;取上清液至另一EP管中,加入氯仿500 μL震荡混匀,12 000 r/min 4 ℃离心3 min,取上清,置冰上待测。
1.2.6Western blot检测PGC-1α、SIRT1和cleaved-Caspase-3蛋白水平 从各组细胞中提取蛋白质并转移到膜上,4 ℃下与PGC-1α(proteintech, 1 ∶1 000)、抗SIRT1(proteintech, 1 ∶1 000)、抗cleaved-Caspase-3(proteintech, 1 ∶1 000)以及鼠抗GAPDH蛋白(proteintech, 1 ∶3 000)单克隆抗体孵育过夜;用含有0.1%Tween-20(TBST)的Tris缓冲液重复洗涤膜,用Tris缓冲液(TBS)洗涤1次,二抗孵育1 h;加入超敏ECL化学发光试剂盒显现免疫染色的蛋白质。通过ImageJ软件分析条带强度。
1.3 统计学分析
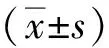
2 结果
2.1 ROS水平
NRK52E细胞培养48 h后,与NG组比较,HG组ROS的表达明显增加,差异有统计学意义(P<0.01)。见图1。
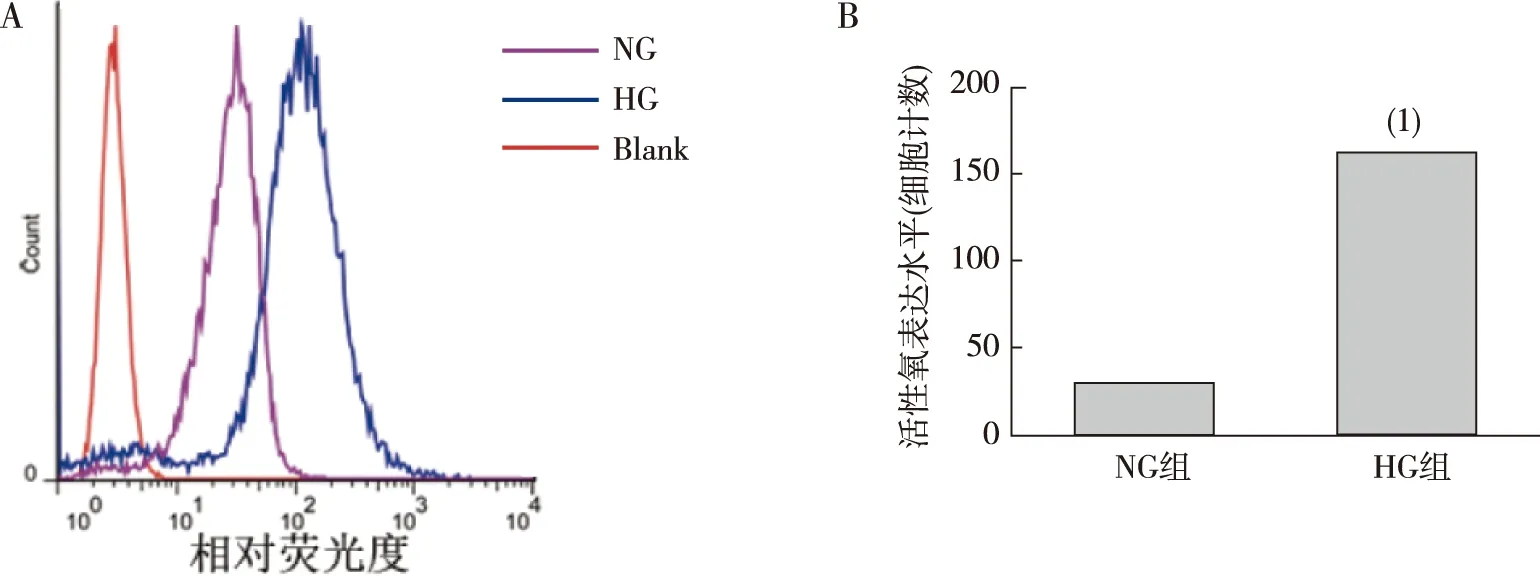
注:A为活性氧的表达水平,B为活性氧表达水平统计;(1)与NG组比较,P<0.01。图1 NG、HG组NRK52E细胞的ROS水平Fig.1 ROS levels of NRK52E cells in NG and HG groups
2.2 ΔΨm表达
NRK52E细胞培养48 h后,与NG组比较,HG组线粒体ΔΨm的表达明显降低,差异有统计学意义(P<0.01)。见图2。
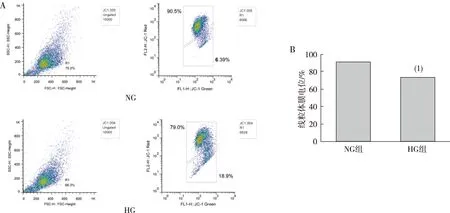
注:A为线粒体膜电位,B为线粒体膜电位统计条图;(1) 与NG组比较,P<0.05。图2 NG、HG组NRK52E细胞中ΔΨm Fig.2 ΔΨm levels of NRK52E cells in NG and HG groups
2.3 线粒体ATP水平
NRK52E细胞培养48 h后,与NG组比较,HG组线粒体ATP水平明显增加,差异有统计学意义(P<0.01)。见图3。
2.4 PGC-1α、SIRT1与cleaved-Caspase-3蛋白的表达
NRK52E细胞培养48 h后,与NG组比较,HG组NRK52E细胞中PGC-1α蛋白表达明显降低(P<0.01)、SIRT1蛋白表达降低(P<0.05),cleaved-Caspase-3蛋白表达明显增加(P<0.01)。见图4。
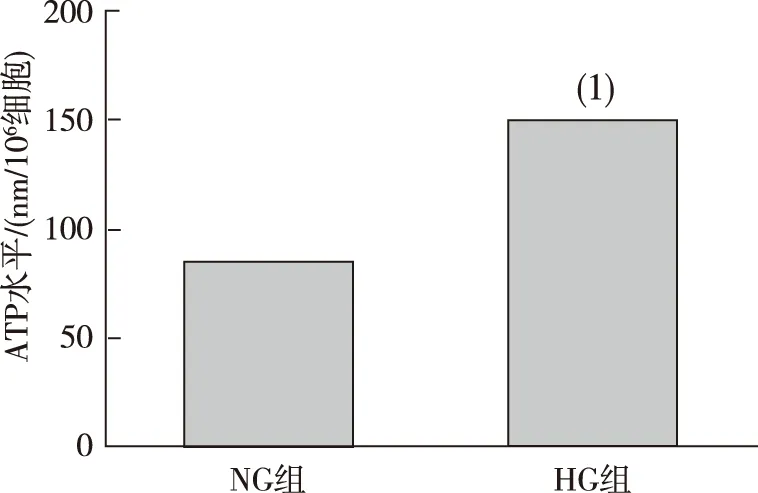
注:(1) 与NG组比较,P<0.01。图3 NG和HG刺激后NRK52E细胞中ATP水平Fig.3 ATP levels of NRK52E cells in NG and HG groups
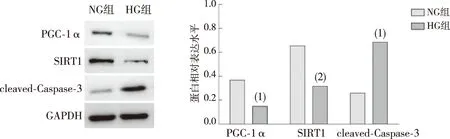
注:与NG组比较,(1) P<0.01, (2) P<0.05。图4 NG、HG组NRK52E细胞中PGC-1α、SIRT1与cleaved-Caspase-3蛋白的表达(Western blot)Fig.4 The protein expression levels of PGC-1α,SIRT1, and cleaved-Caspase-3 in NRK52E cells in NG and HG groups (Western blot)
3 讨论
大量的研究表明,氧化应激在DKD的发生发展中起着独立作用[5-6]。线粒体是ROS产生的主要来源。在DKD进程中,代谢底物的传递对于ATP的产生会改变脂肪酸和氧的含量,糖尿病患者为满足ATP需求而使用的燃料源会导致耗氧量增加,线粒体能量障碍,释放过多的ROS,导致氧化应激,进而从线粒体膜释放细胞色素酶C,激活caspase-9和caspase-3通路,从而引起细胞凋亡和线粒体损伤[6-7]。肾脏组织富含线粒体,具有高能量需求,因此线粒体功能失调可能导致肾固有细胞损伤,导致肾功能和结构损伤。ROS的产生水平超过了局部抗氧化能力,是糖尿病肾脏线粒体功能障碍的生物标志[8]。然而,高糖如何引起线粒体损伤及细胞凋亡的机制不完全清楚。
PGC-1α对于维持能量稳态至关重要。PGC-1α调节线粒体生物发生、肝脏糖异生,参与呼吸链亚单位,包括β-ATP合酶、细胞色素C氧化酶(COX)IV和细胞色素C的调控[9]。早期研究发现,PGC-1α过度表达可减少胰岛B细胞胰岛素分泌,而且PGC-1α在B细胞凋亡、再生、胰岛素分泌以及线粒体新陈代谢中发挥作用[10]。线粒体是一种复杂的细胞器,可以调节多种生理功能的细胞过程和功能,包括氧化还原调节与细胞凋亡等。在DKD的发展过程中,氧化应激是一个主要的病理过程,而川芎嗪硝酮可通过调控PGC-1α的表达进而改善HK-2中线粒体功能,恢复肾脏功能[11]。一方面高糖环境刺激足细胞使线粒体的电子传递链激活,产生氧化应激,大量ROS产生,线粒体功能障碍,另一方面线粒体在胰岛素分泌中起着重要作用。线粒体DNA(mtDNA)突变与糖尿病发病的病因有关[12]。本研究结果也表明,高糖环境下,NRK52E细胞中PGC-1α的表达减少,线粒体膜电位降低,ROS产生增加,凋亡相关蛋白cleaved-Caspase-3蛋白表达增加,这些结果提示,高糖诱导肾小管上皮细胞凋亡、氧化应激,而PGC-1α具有保护线粒体的作用,高糖可能通过抑制PGC-1α的表达引起线粒体功能障碍。
Sirtuins参与抗氧化和氧化应激相关的过程和功能,如线粒体功能、DNA损伤修复、代谢,并影响细胞能量和氧化还原平衡。此外,由于sirtuins的脱乙酰化活性依赖于NAD+,即一种氧化还原信号分子,Sirtuins通过调控控制细胞内NAD+/NADH比率的抗氧化酶和转录因子,在细胞凋亡中发挥作用[13]。SIRT1表达降低,通过去乙酰化FOXO3a活性致氧化应激损伤。线粒体是人体细胞的能量库,并认识到其在ATP和NAD+生产中的关键作用。高血糖症通过电子传递链增加还原当量的穿梭,从而解除对线粒体功能的调控,并导致显著升高活性氧生成和ATP输出减少,增强线粒体ROS的产生,损害线粒体导致功能失调的线粒体积累,引起细胞凋亡[14]。本研究结果观察到,高糖刺激肾小管上皮细胞发生凋亡,凋亡相关蛋白cleaved-Caspase-3蛋白表达增加。然而,高糖刺激肾小管上皮细胞后,细胞发生凋亡的机制不甚清楚。同时也观察到,高糖影响NRK52E细胞,导致PGC-1α与SIRT1的表达降低,ATP的能量供给增加,线粒体膜电位降低,凋亡相关蛋白cleaved-caspase-3增加,结果表明高糖可能通过抑制PGC-1α表达,进而PGC-1α通过抑制下游的SIRT1的表达,诱导细胞凋亡发生。有研究报道,在心肌原代细胞中,PGC-1α 可显著上调 SIRT3 的表达,并提高 SIRT3 的去乙酰化酶活性,PGC-1α 可能通过上调 SIRT3 发挥心肌保护作用[15]。 在高糖处理人肾小管上皮细胞中,通过PGC-1α下调可引起足细胞凋亡[16]。可见,高糖刺激肾小管上皮细胞,PGC-1α可能通过SIRT3介导了细胞的凋亡。具体的机制需要体外干预实验进一步验证。
在DKD的早期阶段,为获得机体所需要的能量,ATP适应性的增加[17],而在糖尿病诱导后4周,伴随着线粒体断裂,线粒体损伤逐渐增加,肾组织ATP含量却逐渐减少[18]。高糖环境,通过氧化应激及细胞凋亡致线粒体功能障碍[19]。其中多种信号通过参与其调控,有研究提到,Akt活化与线粒体功能、氧化应激和凋亡有关,GSK3β是Akt下游的一种蛋白质,广泛参与线粒体功能的调节。Akt被磷酸化激活,激活的Akt(p-Akt)可反向调节GSK3β。在糖尿病肾病,Akt磷酸化被抑制,GSK-3β被激活。然后,活化的GSK-3β调节Bax/Bcl-2的比例,使线粒体通透性增加,刺激线粒体转换孔的开放,促进线粒体释放细胞色素C,最终参与细胞凋亡调节,增加细胞凋亡[20]。在AKI模型的进一步研究发现,通过激活PGC-1α调控SIRT1而抑制氧化应激,可见,PGC-1α与SIRT1在AKI氧化应激中扮演重要角色[21]。在DKD中,腺苷一磷酸活化蛋白激酶(AMPK)是调节PGC-1α信号的一个重要调节器,其磷酸化形式可通过激活下游靶SIRT1而启动PGC-1α信号级联[22]。SIRT1是一种NAD+依赖的组蛋白去乙酰化酶,SIRT1具有多种生物学功能,包括抗衰老、改善糖脂代谢以及胰岛素敏感性的调节。PGC-1α是SIRT1的底物,即SIRT1脱乙酰化PGC-1α,从而提高PGC-1α转录活性[23]。
综上所述,SIRT1和PGC-1α可通过维护线粒体功能,PGC-1α可能上调 SIRT3 的表达,通过Akt通路进而通过减少高糖引起的NRK52E细胞凋亡损伤,对NRK52E细胞具有保护作用。
