材料基因工程加速新材料设计与研发
2022-10-13孙志梅王冠杰张烜广周健
孙志梅 王冠杰 张烜广 周健
(1. 北京航空航天大学 材料科学与工程学院, 北京 100083;2. 北京航空航天大学 集成电路科学与工程学院, 北京 100083)
先进材料是国家工业的支柱,在传统“试错-纠错”研发模式下,材料学家基于自身知识储备和认知能力,通过反复迭代的试错-纠错改进材料性能,实现新材料的设计与研发。 随着新一轮工业革命和互联网时代的到来,新材料的研发速度严重滞后于材料性能需求速度,按需逆向设计和精准控制性能已成为新材料设计的必然趋势。 20世纪末,美国兴起组合材料学(combinatorial materials science, CMS)[1],通过并行合成和高通量表征技术,实现了新材料的快速制备和筛选;21 世纪初,美国和欧洲部分国家提出的集成计算材料工程(integrated computational materials engineering, ICME)[2]将不同时间尺度和空间尺度的多种材料模拟方法相结合,在新材料设计领域取得了突破。 如今,随着计算机和人工智能技术的飞速发展,材料基因工程(materials genome engineering,MGE)被视为实现材料科学技术飞跃和新材料高效研发与设计的基础,是新材料研发的加速器。
材料基因工程是受人类基因组计划(human genome project, HGP)的启发而建立的。 在生物学中,基因是一组编码信息,被视为生物体生长和发育的蓝图,而在材料领域,基因可被看作决定其宏观性能的微观特征单元。 作为基于数据驱动的科学发展第四范式,材料基因工程将高通量计算和设计、高通量制备、高通量表征、材料数据库和人工智能相结合,大大缩短了材料研发周期、降低了研发成本,从而快速研发出满足日益增长的性能需求的新材料。
2002 年,美国宾夕法尼亚州立大学Liu[3-4]首次提出材料基因的概念,并于2005 年设立材料基因基金会。 2011 年,美国正式发布提升美国全球竞争力的材料基因组计划(materials genome initiative, MGI)[5],确立了面向未来的集成计算、实验和数据库的材料研发新模式。 MGI 构建了包含上百万条先进能源材料的计算模拟结果材料数据库,并提出了计算机辅助材料研发、模块化的材料模拟体系、开放式的材料高性能数据库以及多尺度计算融合等研究方向。 欧盟也相继提出了“新材料发现NOMAD”计划、德国推出了工业4.0战略、俄罗斯推出“2030 年前材料与技术发展战略”、中国提出了“材料基因工程”等一系列政策将新材料探索和材料创新设计与研发作为首要发展目标。
本文首先介绍了国内外材料基因工程领域常用的高通量计算模拟软件和框架。 其次,从材料数据来源、多类型数据库和数据标准方面介绍了目前常用的材料数据库。 然后,总结了机器学习方法在材料学中的热点应用,重点介绍了笔者团队自主开发的多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE 研究进展。 最后,总结提出了材料基因工程未来的重点发展方向。
1 材料高通量计算方法
科学发展经历了如图1 所示的4 个过程:从文艺复兴时期实验主导的经验范式、以经典力学和热力学为主导的理论模型范式、基于分子动力学和密度泛函理论的计算科学范式,到如今的大数据驱动的科学研究范式[6]。 近年来,美国、欧洲、日本和中国等国家科研人员在数据驱动研发模式的推动下,开发了一系列用于材料计算基础设施的高通量计算框架和“即插即用”功能完善的高通量计算软件,如表1 所示。 2011 年,在美国国家科学技术委员会、能源部和教育部支持下,加利福尼亚大学伯克利分校劳伦斯伯克利国家实验室Jain 等[7]主导开展了材料基因组项目(materials project, MP),该项目开发了4 款分别用于材料建模、材料计算模拟、自动纠错和服务器部署的高通量计算分析软件。 其中,FireWorks 用于构建材料高通量计算模拟框架,解决高通量材料计算过程中多任务间的依赖关系和任务间的参数及数据传递;Custodian 用于高通量计算过程中的自动纠错;Pymatgen 通过抽象的Python 对象解析材料结构对称性,自动分析不同尺度计算模拟软件的输入参数和结果,工作原理如图2 所示;Atomate则实现了完整的高通量流程及数据存储和服务器的配置,如图3 中能带计算工作流所示。 该项目以其独特的命令行操作方式为高通量计算奠定了软件基础。
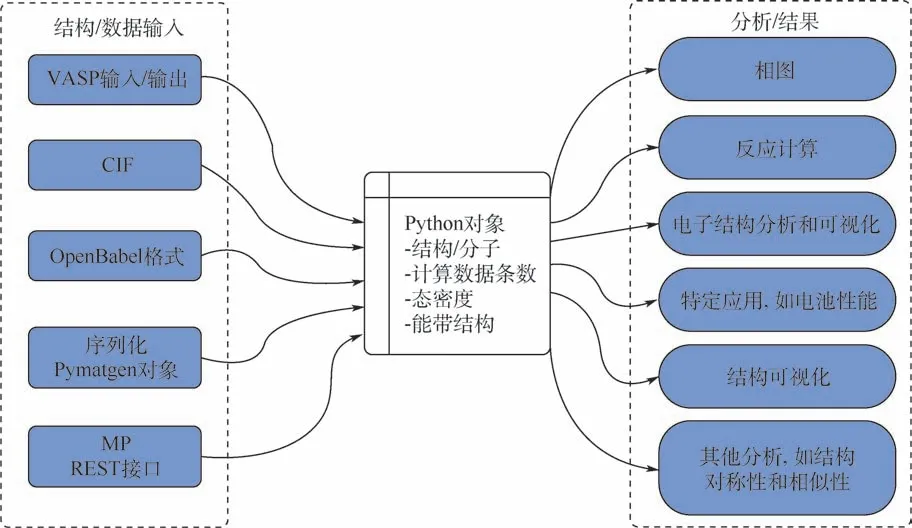
图2 Pymatgen 高通量软件工作原理[7]Fig.2 Principle of Pymatgen high-throughput software[7]
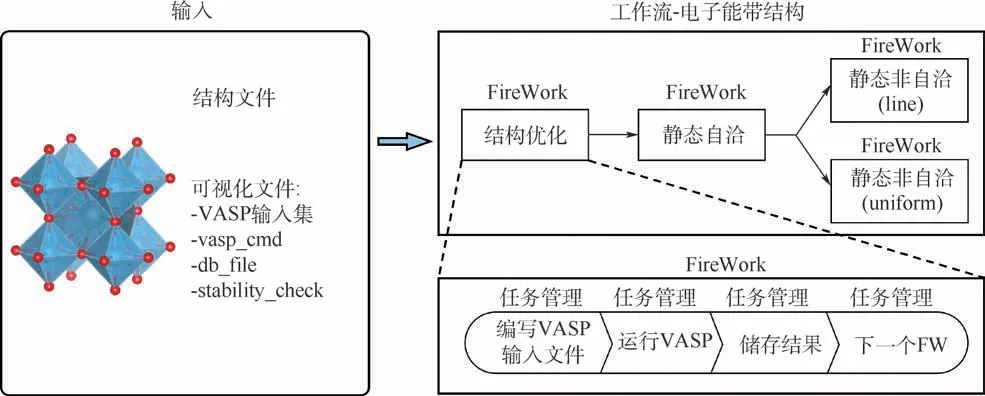
图3 Atomate 中高通量能带计算流程[7]Fig.3 High-throughput computational workflow of band structure in Atomate[7]

表1 材料高通计算软件和框架发展现状Table 1 Software and frameworks of material high-throughput calculation
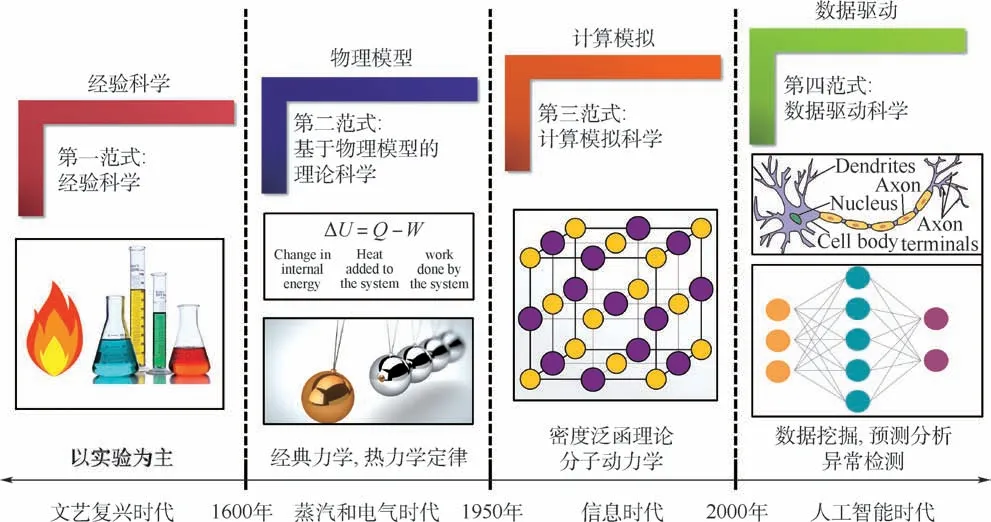
图1 科学发展的四个范式[6]Fig.1 Four paradigms of science[6]
2012 年,同属美国材料基因组项目的杜克大学Curtarolo 等[8]基于Python2 开发了适用于第一性原理计算的高通量计算软件AFLOW-π。 该软件针对高通量第一性原理计算,集成了数据实时反馈、错误控制、数据管理和归档等功能,可用于实现能带结构、态密度、声子色散、弹性特性、复介电常数、扩散系数等高通量计算,并针对性地优化了紧束缚的哈密顿量(tight-binding Hamiltonians,TBH)计算和数据分析流程。
丹麦科技大学Larsen 团队[9]开发了材料批量化计算平台原子模拟环境(atom simulation environment, ASE), 该软件由于缺少工作流程、计算参数和结果的自动纠错功能,并非完整意义上的高通量计算。 而随着版本的迭代,科研人员为其进一步添加了可视化的用户界面、多个材料软件计算器、多种算法的分子动力学计算软件和多种晶体结构优化算法及边界条件,可以满足不同用户不同功能的计算需求。 目前,该平台包含了ABINIT、CASTEP、CP2K、VASP 和LAMMPS 等常用的材料计算模拟软件。
2016 年,瑞士洛桑联邦理工大学Pizzi 等[10]开发了高通量计算引擎(automated interactive infrastructure and database for computational science,AiiDA),该软件系统基于自动化、数据库和开源共享理念,开发了支持数万个材料计算任务并发运行的高通量算法。 材料科学家不仅关注计算模拟的输入和输出,更关注计算模拟过程中的精度及构型的变化是否准确,因此,该软件保存了材料高通量计算中的子任务依赖关系,并自动跟踪记录所有计算和工作流程的输入、输出和中间元数据,以便在其开放式数据库Materials Cloud 中查询数据。
基于MP 发展的材料高通量计算基础框架,比利时天主教鲁汶大学Gonze 等[11]开发了基于多体微扰论的第一性原理高通量计算软件Abipy;美国佛罗里达大学Mathew 等[12]发展了针对二维材料表面和异质结的高通量计算流程MPInterfaces(见图4);英国伦敦国王学院Lambert等[13]发展了用于原子晶界的高通量计算框架Imeall 等。

图4 二维材料表面和异质结的高通量计算软件MPInterfaces 工作流程[12]Fig.4 Workflow for 2D material surfaces and heterojunctions in MPInterfaces software[12]
相比于国外材料基因工程的研究成果,中国高通量计算起步较晚,但在2016 年《中华人民共和国国民经济和社会发展第十三个五年规划纲要》中材料基因国家重点专项支持下,中国也涌现了多个成熟的材料高通量计算框架和软件。
多尺度集成可视化的高通量自动计算和数据管理智能平台(artificial learning and knowledge enhanced materials informatics engineering, ALKEMIE)是由笔者团队Wang 等[14-15]基于Python 开源框架自主开发的中国第一个高通量自动流程可视化计算和数据管理智能平台。 该平台从设计出发吸取国外材料基因相关软件的先进理念,克服了计算过程中可能遇到的兼容性差、接口不统一和功能拓展困难等问题,开发了包含材料高通量自动计算模拟、材料数据库及数据管理、基于人工智能和机器学习的材料数据挖掘3 个核心理念的智能平台。 ALKEMIE 平台适用于数据驱动的材料研发,详细内容见第4 节。
MIP(materials information platform)是由上海大学Yang 等[16]开发的适用于热电材料高通量筛选的高通量计算软件;MatCloud 是由北京迈高材云科技有限公司Yang 等[17]开发的第一性原理计算引擎,目前支持VASP 和ABINIT 等第一性原理高通量计算软件;JAMIP 是由吉林大学Zhao等[18]开发的开源高通量集成软件,该软件利用人工智能算法在高通量计算的海量材料数据中智能寻求新材料和新原理。 中国材料基因工程高通量计算平台(CNMGE)则是由国家超级计算天津中心开发的网页版高通量集成计算平台,该平台可以集成多种不同的高通量计算软件,包括ALKEMIE 高通量智算平台、含能材料分子专用高通量筛选系统EM-Studio 和用于无机骨架材料的晶体结构解析与预测软件(framework generator, FraGen)。
上述高通量计算软件和框架,一方面为科研人员提供了快捷方便的自动计算工作流,可以在高性能超算中实现高效并行计算,显著提升计算效率;另一方面避免了传统试错-纠错法中可能出现的人为误差,使得研究人员有更多的精力关注材料科学问题本身,而非高通量所涉及的技术难题。 近年来,高通量计算模拟已经在材料构型预测、材料结构稳定性和相稳定性预测、能源材料能量转化效率、最优掺杂元素分级筛选等方面获得了广泛应用。 举例来说,Curtarolo 等[19-20]通过高通量方法从435 个含d 电子的二元金属间化合物中筛选出了283 个能量稳定化合物,其中273 个(96.5%)化合物最终获得了实验验证;Hu 等[21]基于第一原理计算的层级筛选,从29 个过渡金属元素中筛选出可以提高相变材料Sb2Te3性能的最佳掺杂元素Y、Sc 和Hg,除了有毒的Hg 元素,Y 和Sc 均获得了实验验证;Gan 等[22]基于第一原理计算的高通量分级筛选,通过结构能量、声子谱、力学稳定性和转换效率等4 个筛选标准从21 060个候选光电材料中筛选出了78 个稳定化合物,且其中22 个化合物的性能优于当前太阳能电池材料GaAs。
2 材料数据库研究进展
高通量计算大幅提高了计算模拟的效率,并产生了海量的数据,这些数据中既包含了有用的材料性质数据,又包含了大量重复的无效数据。而材料学中由于所研究的材料体系、成分、结构等的不同,材料测试、制备工艺和流程也不尽相同。对于不同用途材料,所关注的材料性能和关键指标也有差异,因此,在数据库的构建过程中面临数据存储类型、数据库的兼容性和泛化能力等一系列问题。 本文根据材料数据来源,将数据大致分为材料计算数据、材料测试数据和已发表的文本数据三大类。 本节将介绍目前国内外常用的材料数据库和数据共享标准。
2.1 材料多类型数据库
目前常用的材料数据库如表2 所示。 ICSD是由莱布尼茨信息基础设施研究所Belsky 等[23]构建的无机材料结构数据库,收录了自1913 年以来在1 600 个学术期刊发表的超过8 万篇论文中的共计21 万条晶体结构数据,覆盖金属、有机物、同素异形体等各种形态的材料体系;COD (crystallography open database) 是由英国剑桥大学Quirós 等[24]开发的包含超过700 万个有机、无机、金属有机化合物和矿物的晶体结构数据库;MP Database 是由美国加利福尼亚大学伯克利分校劳伦斯伯克利国家实验室Jain 等[7]建立的材料计算模拟专用数据平台(见图5),不仅收录了材料结构数据,也收录了元素性质、电子结构、弹性张量和能源转换电极性能等数据;AFLOWLIB(automatic flow lib)是由美国杜克大学Curtarolo等[19]基于AFLOW-π 高通量软件开发的材料计算数据库,数据库中收录了6 400 余条热力学相图数据和超过45 万个四元混合物的材料性质数据;Materials Cloud 是由瑞士洛桑联邦理工大学Pizzi 等[10]开发的第一性原理计算元数据的数据库,包括超过752 万条第一性原理结构弛豫流程和纳米多孔材料吸收和扩散相关的材料性质数据;OQDM(open quantum database for materials)是由美国西北大学Saal 等[25]开发的第一性原理计算热力学数据库,包含了数万个二元、三元和四元相图;NOMAD(novel materials discovery)是由欧洲马克斯·普朗克学会Draxl 等[26]开发的欧洲最大的新材料共享数据库,该数据库包含了49 TB的各类材料数据;MatNavi 是由日本国家材料科学研究所Ogata 等[27]开发的多种材料数据的集合,包含聚合物数据库(化学结构、加工、物理性质、NMR 光谱数据)、无机材料数据库(晶体结构、相图、物理性质)、金属材料数据库(密度、弹性模量、蠕变性质、疲劳特性)、电子结构计算数据库等。

图5 Materials Project Database 数据库概况[7]Fig.5 Snapshot of Materials Project Database[7]

表2 材料多类型数据库的发展Table 2 Development of multi-type databases of materials
除了上述通用的多类型材料数据库,还有众多针对材料某个特定领域的特色数据库。 例如,美国佛罗里达大学开发的材料表面界面数据库Materials Web; 美国国家标准技术研究所Choudhary 等[28]开发的赝势数据库JARVIS-DFT;丹麦科技大学Larsen 等[9]开发的二维材料、硒化物和硫化物数据库(computational materials repository, CMR);北欧理论物理研究所Borysov 等[29]开发的三维有机晶体材料电子结构和带隙数据库(organic materials database,OMDB);美国斯坦福大学Hummelshøj 等[30]开发的催化材料活化能数据库CatApp;此外,还有ASM 出版社开发的热处理数据表及应力应变和蠕变曲线材料数据库(ASM alloy center database,ASMDB)、矿物材料数据库(American mineralogist crystal structure database,AMSD)和储氢材料数据库(hydrogen storage materials database,HSMD)等。
中国目前也发展了多个大型材料数据库共享平台。 ALKEMIE-DB 是由笔者团队Wang 等[14-15]基于高通量智能计算平台ALKEMIE 开发的多类型材料数据库(见图6),该数据库分为隐私数据库和共享数据库两大类,根据数据类型进一步细分为含60 余万组数据的晶体结构数据库、含1 万余条声子能带的声子谱数据库、含20 余万组数据的深度学习赝势数据库、高通量计算工作流数据库等。 MSDSN 是由国家统筹建设、北京科技大学实施完成的材料科学数据共享平台,主要分为包含有色金属材料和特种合金和微观组织模拟的实验数据库、热力学和动力学相关的计算模拟数据库等。 Atomly 是由中国科学院物理研究所松山湖材料实验室和怀柔材料基因平台共建的材料计算数据库,目前包含20 万条材料结构数据和5 万条相图数据等。 MCDC(national materials corrosion and protection data center)是由国家科技部门建设的腐蚀防护数据平台,包含环境数据、腐蚀数据、腐蚀检测和腐蚀预测等。

图6 ALKEMIE-DB 数据库中高通量能带和态密度计算结果可视化[14]Fig.6 Visualization of high-throughput band structures and density of states calculations in ALKEMIE-DB databases[14]
2.2 材料数据标准
随着高通量计算、高通量实验和超级计算机计算能力的发展,材料数据形成的数据海(data ocean)面临着5 个重要的挑战(“5V”特性):
1) 速率(velocity)。 新数据产生速率和旧数据更新速率越来越快,对数据的格式化存储和快速读写提出了更高的要求。
2) 数据量(volume)。 材料数据以TB 量级不断增加,需要可靠的数据存储、高效的数据检索和开放共享的可重复利用。
3) 多样性(variety)。 材料数据的存储形式、材料体系、测试和计算方法,以及数据蕴含的物理意义更加多样化。
4) 真实性(veracity)。 数据的不确定性和可靠性决定了数据是否真实有效,进一步决定了数据挖掘和机器学习模型的精度和泛化能力。
5) 低价值密度(value)。 数据价值密度高低与数据总量大小成反比,数据量越大,数据价值密度越低。
如何在海量数据中分析预测数据隐藏的真实意义和价值是材料基因工程方法努力探索的主要方向。
为了解决上述问题,欧洲马克斯·普朗克学会Draxl 等[26]提出了材料数据库建设的FAIR Data 准则,即可发现(findable)、可获取(accessible)、可互操作(interoperable)和可再利用(reusable),来提升材料数据的开源共享性。 欧洲用于材料设计的开放式数据库集成团队Andersen 等[31]提出了材料数据共享标准OPTIMADE(见图7),该标准通过JSON 格式定义了材料数据共享的统一标准,目前大多数材料数据库MP Database、AFLOWLIB、NOMAD、Materials Cloud 和ALKEMIEDB 等均提供了OPTIMADE 通用接口支持。
图7 材料数据共享标准OPTIMADE 概况[31]Fig.7 Overview of materials data sharing standard of OPTIMADE[31]
中国中关村材料试验技术联盟于2019 年也提出了材料基因工程数据通则,将材料科学数据分为样品信息、源数据(未经处理的数据)与衍生数据(经分析处理得到的数据)三大类,并从宏观上定义了材料数据的通用性与专用性;北京科技大学材料基因工程北京市重点实验室在2020 年MSDSN 数据库中定义了材料科研数据DOI 编码规则:“10. 11961/classification. project. date. sequence”,编码中包含了材料数据分类号、项目支撑信息、注册日期和流水号;北京航空航天大学ALKEMIE-DB 数据库也发展了用于材料数据共享的唯一标识符:“alkemie. date. classification/user_defined_label. number.”,其中alkemie 为数据库社区唯一标识,date 代表数据创建日期(精确到μs),classification 表示数据类别,user_defined_label 为用户自定义字段,number 为数据唯一索引序号。
3 机器学习在材料设计中的应用
机器学习传统上分为监督学习和非监督学习两大类。 监督学习是指给算法一个数据集,对于数据集中的每一个样本,都给出对应的映射(即标签),算法的目的是给出更多的映射,得到更多的答案;而非监督学习常常被用于在大量无标签数据中探索并发现规律。 进一步,监督学习分为处理连续值的回归问题和处理离散值的分类问题。 目前,常用的机器学习算法已经有很多相关综述,本文不再赘述。 随着计算机科学和人工智能的发展,机器学习在材料结构设计、材料性能预测和材料分析图像识别等领域扮演着越来越重要的角色。 本节主要介绍机器学习在材料性能预测、材料数据文本挖掘和机器学习原子间作用势。
3.1 材料性能预测
机器学习在材料学中的应用主要包含格式化材料数据、机器学习模型训练和高效材料性能预测3 个步骤,并在二维、光伏、催化、合金和热电等材料中均获得了显著的研究成果。 近年来,瑞士洛桑联邦理工大学Lin 等[32]通过高通量筛选和机器学习算法,从ICSD 和COD 数据库的108 423个三维晶体构型中,通过对称性和几何算法筛选了1 036 个容易合成和789 个具有潜在可能性的层状二维材料混合物;东南大学Lu 等[33]通过高通量筛选和机器学习描述符,从5 158 个候选材料中筛选了能量转化效率高且带隙在0.9 ~1.6 eV之间有机无机结合的钙钛矿光伏材料(HOIPS),并构建了可以高效预测带隙值的结构描述符和机器学习模型,进一步将模型拓展到双层HOIPS 中,成功从11 370 个混合物中预测了204 个无毒且稳定的光伏材料[34];北京科技大学Zhang 等[35]在铜合金体系中通过贝叶斯优化迭代方法,分别构建了误差小于7%的硬度模型和误差小于9%的电导率模型,并通过迭代优化设计了兼具优异力学和电学性能的Cu-Ni-Co-Si-Mg 合金。
3.2 材料数据文本挖掘
2018 年,瑞士联邦理工学院Villars 等[36]通过计算机视觉和自然语言方法从已经正式发表的论文中自动识别有效材料结构和数据,并通过数据挖掘探索数据背后隐藏的物理模型和机理,构建了MPDS(materials platform for data science),近年来,该团队首次从35 000 篇论文中解析了15 500个化学成分,并基于其中的2 330 个二元体系构建了机器学习分类模型;进一步从超过8 000 篇发表的论文中构建了2 800 个二元混合物的原子配位环境多面体分析算法(atomic environment types,AETs);随后,从超过50 000 篇已发表论文中分析了290 000 个原子配位环境数据,将该算法模型拓展到了多元无机混合物中。 2019 年,Tshitoyan 等[37]发展了Word2vec 非监督机器学习模型,成功从330 万材料文本中筛选了超过7 000 个候选的热电材料。 除了材料计算模拟数据,在实验合成方式上,Kononova 等[38]通过文本挖掘和自然语言处理方式从53 538 篇科学文献数据中筛选了19 488 个无机金属合成方式,包括材料成分、制备条件、化学平衡方程和反应过程,该数据库为实验中无机材料的制备过程提供了有力的数据支持。
3.3 机器学习原子间作用势
材料计算模拟根据模拟时长和体系大小分为原子尺度、分子尺度、介观尺度和宏观尺度模拟,尺度越小模拟精度越高,尺度越大越接近真实体系,但是不同的模拟尺度采用的物理模型和近似原理不同,数据耦合非常困难,而数据驱动的机器学习方法被视为材料多尺度模拟的耦合剂。 经典大规模分子动力学常被用来模拟近似真实材料体系的服役性能,但可靠、精确的原子间势函数的匮乏限制了其广泛应用。 基于密度泛函原理(DFT)的第一性原理模拟具有精确的赝势库,但求解本征值所需的巨大计算量限制了该方法在大的原子尺度和时间尺度上的模拟,常用的VASP 仅限数百原子的体系。 因此,简单方便地获得适用于经典分子动力学的可靠势函数非常重要。 随着计算机技术、计算机视觉和材料基因理念的快速发展,通过机器学习结合大数据、高通量计算的方法拟合可靠的适用于经典分子动力学模拟的势函数成为了研究热点。
机器学习势函数的发展主要经历了原子个数受限的低维度势函数和泛化能力强的高维度神经网络势函数2 个发展过程。 1995 年, Blank 等[39]开发了第一个基于统计学的势函数模型,用于研究氢原子的分子动力学模拟;2009 年,Malshe等[40]进一步提出了通过神经网络预测经典多体势方程参数的模型。 但是,上述模型均不能改变输入的原子个数,因此限制了机器学习势函数的应用。
2011 年,Behler[41]提出了原子中心对称函数,通过数学方程解析原子局域环境,构建了输入原子个数不受限的高维度神经网络模型。2018 年,Gastegger 等[42]发展了权重相关的对称函数(wACSF),通过卷积神经网络提升了模型的精度和实用性,但是由于局域近似,无法包含超过截断半径的原子长程相互作用。 2018 年,Yao等[43]提出了包含长程静电作用和散射作用的HDNNP 方法,但是该方法并未获得广泛应用,一方面由于物理学中超过6 ~10 Å 的静电作用通常对体系的整体影响较小,另一方面添加长程作用会急剧增加模型训练成本,与其对精度的微小提升相比有待进一步优化。 笔者团队Wang 等[14]开发了适用于相变材料Sb 单质的跨尺度机器学习势函数PotentialMind,该势函数模型与DFT 比较,对能量预测的精度达到98%,平均到每个原子上的能量误差值小于0.045 eV/atom,对力的预测精度达到89%,该算法具有很强的扩展性和通用性,易于扩展到多元材料体系中。
机器学习势函数方法一方面实现了具有第一原理精度且更大原子数体系和更长时间尺度的大规模分子动力学模拟,另一方面通过替代求解复杂多体薛定谔方程本征值,使得模拟速度提升2 ~3个数量级,目前,该方法已经在多个材料体系中获得了应用。 例如,Sosso 等[44]发展了适用于二元相变存储材料GeTe 的人工神经网络势函数,并实现了具有第一原理精度的4 096 个原子体系的大规模分子动力学模拟,通过模拟相变材料的多个淬火过程(100 ~300 ps),探究了淬火速度和模拟体系大小对GeTe 非晶结构的影响;Artrith和Urban[45]基于Fortran 语言开发了适用于钙钛矿TiO2的神经网络势函数软件(atomic energy network, AENET),并面向科研人员开源使用,加速了机器学习势函数方法在能源材料中的应用;Mocanu 等[46]通过高斯近似方法构建了三元相变材料Ge2Sb2Te5的势函数模型,实现了含7 200个原子的非晶体系的大规模分子动力学模拟,揭示了模型大小和原子个数对非晶局域结构的影响,并阐明了非晶构型中化学键和晶化过程中的微观结构演化;Zhang 等[47]开发了适用于高性能并行计算的深度神经网络势函数方法DeePMD,实现了模拟体系超过1 亿原子、模拟时长超过1 ns 的大规模分子动力学模拟,显著加速了新材料设计与研发。
4 多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE
2016 年,笔者团队在国家重点研发计划材料基因工程专项的支持下,基于Python 开源框架自主开发了一套多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE,主要包含高通量自动工作流ALKEMIE Matter Studio(MS)、数据管理及材料数据库ALKEMIE Database(DB)、基于机器学习的材料数据挖掘ALKEMIE Potential Mind(PM)三部分[14-15]。
4.1 高通量智能计算ALKEMIE-MS
ALKEMIE 基于AMDIV 设计理念,解决了材料高通量智能模拟过程中5 个核心问题:自动化计算(automation)、模块化拓展(modular)、材料数据库(database)、人工智能方法(intelligence)和可视化界面(visualization),可实现从建模、运行到数据分析,全程自动无人工干预。
ALKEMIE 中,多尺度集成的高通量自动计算可以通过不同模块间以搭积木的方式实现自动耦合并完成计算。 不同模块的连接方式如图8 所示。 首先,由建模模块控制输入,通过多种建模方式将材料构型导入高通量预处理器,科学计算模块控制任务的计算顺序和纠错(可进行电子尺度、原子尺度、分子尺度和介观尺度的高通量自动流程)。 然后,服务器用来协调计算资源,配置远程节点,实现本地与远程服务器通信并提交任务,数据存储系统负责保存整个流程中所有的元数据,将计算结果保存在不同类型的数据库中。 最后,通过数据分析和人工智能进行数据挖掘。

图8 多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE 计算模块概况Fig.8 Overview of platform with multi-scale integration of visualized automatic high-throughput calculation and intelligent data management ALKEMIE
目前,该软件可以实现超过104量级的高通量并发计算,包含第一性原理计算VASP、QE 和ABINIT,分子动力学模拟LAMMPS 和ASE,热力学计算软件Gibbs,动态蒙特卡罗模拟(KMC),相场相图模拟OpenPhase 和OpenCalphd 等多尺度模拟软件,可通过参数传递的方式实现跨尺度计算。 目前,该软件系统已部署在9 家超算中心,包括4 家国家超算、4 家高校超算和1 家企业超算。软件可移植性和可拓展性强,适用于对材料模拟掌握程度从初级到专业的所有材料研究人员,可提供材料建模、高通量智能计算、数据挖掘和人工智能一体化的新材料设计方案。
4.2 材料多类型数据库ALKEMIE-DB
ALKEMIE-DB 材料多类型数据库分为五大类:材料结构数据库、工作流源数据库、材料性能数据库、机器学习描述符数据库和论文数据库。目前,收录了超过64 万条材料结构数据、296 条相图数据、1 万条声子能带数据、1 418 条赝势数据和20 余万条机器学习描述符数据。 通过ALKEMIE中JSON 格式的API 和国际通用的OPTIMADE 格式的API 实现数据高效查询检索,并通过Finder 控件实现结构的数据可视化,如图9 所示[14]。

图9 ALKEMIE-DB 材料结构数据库[14]Fig.9 Material structure database of ALKEMIE-DB[14]
4.3 材料学机器学习应用框架ALKEMIE-PM
机器学习在材料学中通常被视为未知的黑盒模型,而材料研究人员相比于机器学习方法更关注材料性能、成分和工艺问题,因此,如何构建简单通用的可视化机器学习框架至关重要。 ALKEMIE 通过抽象凝练高级API 及规范化和格式化机器学习的每个具体步骤开发了一套通用的可视化机器学习的流程。 Datasets 模块给定了数据集及特征的输入格式,Model 模块定义了不同的机器学习算法,Evaluate 模块给出了机器学习训练过程的收敛情况及模型在测试集或模型在部署过程中的应用情况,Plotter 模块中多种分析方法提供了将训练过程及其结果可视化的功能,如图10所示。

图10 ALKEMIE-PM 机器学习框架Fig.10 Framework of ALKEMIE-PM machine learning
目前,笔者团队基于ALKEMIE 可视化机器学习框架发展了一系列高效机器学习模型。Chen 等[48]通过团簇展开法和高通量第一性原理计算搜索了二维过渡金属硫化物的单层和双层无序掺杂结构,得到了稳定掺杂结构和能量之间的对应关系,通过机器学习挖掘了影响半导体-金属转变(SMT)的2 个关键特征,即范德华间隙内掺杂氧原子的浓度差和Mo-S/O 键角正切的平均值(tanθ);Peng 等[49]通过线性回归方法构建了适用于MXene 材料单原子催化剂结构与氧还原反应(ORR)性能构效关系的组分描述符,基于简单的元素性质可以高效快速预测材料的催化活性;Gan 等[50]通过高通量筛选和可视化机器学习框架,开发了2 个精度分别为90.90%和91.67%的机器学习模型预测层状IV-V-VI 族半导体不同温度下的最大热电优值(ZTmax)和实现ZTmax所对应的最佳掺杂类型,并成功从840 个候选成分中筛选出数种具有优良潜力的热电材料。
4.4 未来的发展方向
综上所述,ALKEMIE 已经研发了集可视化高通量自动计算流程、材料多类型数据库和人工智能方法于一体的新材料设计研发智能平台,但是未来仍有亟须发展的新方向和新方法。
在高通量计算方面,开发从原子、分子、介观到器件的跨尺度模拟方法是目前极具挑战且具有广阔应用前景的热点问题。 Martin Karplus、Michael Levitt 和Arieh Warshel 三位科学家在分子领域凭借量子力学和分子动力学跨尺度模拟方法(QM/MM)获得了2013 年诺贝尔化学奖。 而在周期性材料的研究方面,由于体系周期性边界条件和原子局域环境的复杂性,使得跨尺度模拟的精度非常难以控制,发展高通量跨尺度高并发、自动纠错及数据耦合方法,通过机器学习数据挖掘等算法进一步提升跨尺度模拟精度是未来的研究热点之一。
材料数据库方面,应该保持开源和共享的发展理念,基于FAIR 原则,构建包含材料计算和实验元数据及中间数据的高效数据库,发展数据规模更大、种类更丰富且具有航空特色的共享数据平台,完善更加通用兼容的数据标准和共享标识均是未来重要的研究方向。
在机器学习领域,材料学中数据集的构建非常困难,因此,研发基于小数据集的高效机器学习模型训练算法至关重要;由于机器学习模型的黑盒特性,探索可解释的机器学习模型,阐明模型背后隐藏的物理意义,实现逆向材料成分和结构设计也是未来的热门研究领域。
5 结束语
材料基因工程颠覆了传统的“试错-纠错”材料研发模式,通过数据驱动的高通量方法和人工智能模型加速新材料的研发与设计。 本文系统总结了国内外知名的材料高通量计算框架、常用的针对多种材料体系的大型材料数据库和机器学习方法,并概述了多尺度集成可视化的高通量自动计算和数据管理智能平台ALKEMIE 的研究进展,提出了未来发展的研究方向,为实现按需逆向设计新材料提供参考。
