胰高血糖素样肽-1受体敲除H9c2细胞株建立及其抗凋亡作用初探
2022-10-13林筝鸣钱航李东锋许浩陈继舜闵新文陈俊李小雷杨汉东
林筝鸣 钱航 李东锋 许浩 陈继舜 闵新文 陈俊 李小雷 杨汉东
(1.锦州医科大学国药东风总医院研究生培养基地,湖北 十堰 442008;2.湖北医药学院附属国药东风总医院,湖北 十堰 442008)
糖尿病心肌病(diabetic cardiomyopathy,DCM)是糖尿病患者的主要心脏并发症之一,DCM是指在无冠心病、高血压、瓣膜或先天性心脏病的情况下所致的心脏结构和功能异常。虽然DCM定义明确,但其病理生理机制仍待阐明[1-2]。有研究[3]表明,心肌细胞的凋亡在DCM的发展中至关重要。
甲基乙二醛(methylglyoxal,MGO)是晚期糖基化终产物的主要前体,有研究[4-6]表明,MGO促进心肌细胞凋亡及心功能障碍。线粒体在细胞凋亡中发挥着重要作用,在各种刺激下,线粒体膜电位(mitochondrial membrane potential,MMP)下降的同时线粒体膜通透性发生改变,使细胞色素c从线粒体渗漏至细胞质中,促进胱天蛋白酶-3(caspase-3)级联反应,诱导细胞凋亡[7]。有研究[8-9]表明,MGO通过诱导线粒体功能损伤,导致β细胞和肝细胞凋亡。此外,心肌细胞在MGO诱导下线粒体活性氧产生增加,MMP降低,细胞凋亡显著增加[10]。MGO通过乙二醛酶(glyoxalase,GLO)系统解毒,使MGO脱毒为D-乳酸,抑制细胞损伤及凋亡[11]。
胰高血糖素样肽-1受体(glucagon-like peptide-1 receptor,GLP-1R)是B类G蛋白耦联受体家族中的一员。但因为检测手段敏感性和特异性的限制及不同物种之间GLP-1R的表达差异,GLP-1R在心脏的精确定位一直存在争议[12]。有研究表明在人类心脏的四个腔室中均检测到GLP-1R mRNA表达。然而通过原位杂交仅在右心房检测到GLP-1R的存在[13]。与之相悖的是,Clarke等[14]用单克隆抗体3F52免疫组织化学检测到人类心房肌和心室肌中有广泛的GLP-1R免疫阳性细胞。在另一项使用单克隆抗体3F52对人类的心脏GLP-1R定位的试验[15]中表明,窦房结细胞GLP-1R免疫反应阳性。对小鼠心脏GLP-1R表达的分析显示,心室GLP-1R mRNA转录水平远低于心房[16]。在大鼠心脏中,通过免疫组织化学和蛋白质印迹法均检测到了GLP-1R的表达,在缺血再灌注损伤后,GLP-1R的表达水平显著降低[17]。但该实验并未对大鼠心脏GLP-1R的表达进行精确的定位。在众多以H9c2细胞为研究对象的实验中发现,胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)及其类似物能通过抗氧化应激、抑制线粒体功能障碍、改善能量代谢、逆转心肌肥厚等机制,在心肌细胞应对不同的损伤时表现出良好的心肌保护作用[18-21],虽然这些研究并未深入地对H9c2细胞GLP-1R的表达情况进行验证,但GLP-1R激动剂对心肌的保护作用也能验证GLP-1R在H9c2细胞的表达及功能。另有研究[22]表明,GLP-1能有效地提高脂肪组织中GLO1的活性。尽管之前的多项研究已揭示GLP-1R激动剂对心肌细胞的保护作用,但这些作用的机制至今尚未完全阐明。本研究旨在发现GLP-1R在MGO诱导的线粒体功能障碍和心肌细胞凋亡过程中的保护作用。
CRISPR/Cas9是原核生物的获得性免疫系统,能抵御外源遗传物质的入侵,由此发展的基因编辑技术能很好地实现基因的敲除及敲入[23-24]。此外,尚未有研究基于CRISPR/Cas9基因编辑技术敲除H9c2细胞GLP-1R基因研究其生理重要性。因此,本实验将通过CRISPR/Cas9基因编辑技术敲除H9c2细胞的GLP-1R基因,构建GLP-1R基因敲除H9c2细胞稳转株,观察GLP-1R缺乏对H9c2细胞表型的影响,旨在研究GLP-1R蛋白的表达对MGO诱导的H9c2细胞线粒体功能障碍、细胞凋亡的预防作用及通过调节GLO1的解毒作用。
1 材料与方法
1.1 材料
大鼠H9c2心肌细胞(购自中国科学院上海细胞库)、DNA胶回收试剂盒(生工SanPrep)、pSpCas9-puro载体(Addgene)、限制性内切酶(Biolabs)、胶回收试剂盒(Qiagen)、无缝克隆试剂盒(Clontech)、质粒提取试剂盒(Qiagen)、嘌呤霉素(Ubigene Bioscience Co.Ltd.,Guangzhou,China)、MGO(M0252,Sigma)、GLP-1R抗体(26196-1-AP,Proteintech)、GAPDH抗体(AntGene)、二抗山羊抗兔(AntGene)、CCK-8试剂盒(C0005,Targetmol)、F-actin(C2207S,Beyotime)、Hoechst 33342活细胞染色液(C1028,碧云天)、JC-1试剂盒(碧云天)、RNasin®RNA酶抑制剂(N251A,Promega)、M-MLV(M170B,Promega)、M-MLV×5(M531A,Promega)、GoTaq®qPCR(A600A,Promega)、dNTP(上海生工)、Oligdt(上海生工)、实时荧光定量PCR引物合成(Sangon Biotech)。
1.2 细胞培养和细胞活力测定
大鼠H9c2心肌细胞在含10%胎牛血清的DMEM培养液和37 ℃、5%的CO2的恒温箱中培养。当细胞密度为80%~90%时按1:3进行传代。用CCK-8试剂测定细胞活力。将GLP-1R-/-组和野生型(wild-type,WT)组的H9c2细胞分别接种于96孔板(1×104个/孔),培养24 h后更换为含有1% FBS的DMEM培养基饥饿处理16 h。将不同浓度的MGO(0~100 μmol/L,用PBS稀释)处理细胞24 h,或用50 μmol/L MGO处理细胞不同时长(0~48 h)。酶标仪读取吸光值,计算细胞活力。
1.3 靶位点及鉴定引物设计
使用在线CRISPR设计工具(https://en.rc-crispr.com/)网站,搜索GLP-1R基因(ID:25051)信息。以GLP-1R基因2号外显子为靶点分别在其外侧及内部设计4条向导RNA(guide RNA,gRNA)(图1)。在该基因的外显子序列中寻找PAM序列(NGG),选取该序列上游紧邻的20 bp碱基序列作为gRNA,最终选择4条gRNA:gRNA1:5’-GGATTGCACAAGGTTGCCGTGGG-3’、gRNA2:5’-GCTCCCGCACCCCTCCAAAGAGG-3’、gRNA3:5’-GGACACCGTGGCACCCTATGAGG-3’和gRNA4:5’-TGAAGTGTAGCGTAGTCACCAGG-3’,同时设计靶序列的鉴定引物(图1和表1)。

图1 GLP-1R gRNA靶位点突变鉴定引物

表1 GLP-1R gRNA靶位点突变鉴定引物序列
1.4 目的基因片段获得
采用PCR产物体外转录的方法合成gRNA。PCR产物进行琼脂糖凝胶电泳检测扩增效果,琼脂糖凝胶电泳后,用生工的SanPrep柱式DNA胶回收试剂盒做胶回收。
1.5 目的基因构建入线性化载体中
用BbsI酶切pSpCas9-puro载体,酶切产物进行琼脂糖凝胶电泳检测酶切效果,并把目的载体条带从琼脂糖凝胶电泳后的胶中切割下来,用QIAquick Gel Extraction Kit(Qiagen)做胶回收。使用无缝克隆试剂盒,将目的基因片段和线性化载体加到离心管中进行重组反应,转化至E.coli Stbl3 感受态细胞中,把转化物涂到LB平板上,孵育过夜,挑取转化子重悬,行菌落PCR,鉴定阳性克隆后提取质粒,并将质粒送测序公司进行测序验证。
1.6 质粒的电转和基因敲除细胞系的筛选
当H9c2细胞密度为80%时,以1 600 V、10 ms、1 pulse电转模式进行电转,回收质粒稀释液,加入完全培养基培养48 h,加入嘌呤霉素至终浓度为2 μg/mL,10 d后挑取单克隆至96孔板,用胰酶消化单克隆阳性细胞,单克隆细胞株培养后,按照基因组提取试剂盒说明书提取基因组DNA,用相应的鉴定引物进行PCR反应,扩增产物送测序。
1.7 免疫印迹
用免疫印迹法检测GLP-1R蛋白表达情况。用培养皿分别培养GLP-1R-/-组及WT组H9c2细胞,当细胞密度为80%时收集细胞,提取各组细胞的总蛋白,BCA法测定蛋白浓度。经10% SDS-PAGE电泳、转膜(聚偏氟乙烯),加入一抗GLP-1R(1:1 000)/GADPH (1:5 000)4 ℃ 孵育过夜,二抗山羊抗兔(1:10 000),室温孵育1 h,TBST洗3次,ECL显色,用凝胶图像分析系统进行扫描分析,实验共进行3次。
1.8 细胞骨架染色和MMP测量
用F-actin对细胞骨架进行染色。将WT组和GLP-1R-/-组H9c2心肌细胞(1×104个/孔)接种于底部放置有盖玻片的6孔板中,24 h后用F-actin进行细胞骨架染色,Hoechst 33342进行细胞核染色,具体方法参考试剂盒说明书。用JC-1荧光法测定MMP。将GLP-1R-/-和WT组H9c2细胞分别接种于放置有盖玻片的6孔板(2×105个/孔)24 h,饥饿处理16 h,用MGO(0 μmol/L,50 μmol/L)刺激1 h后用JC-1进行线粒体染色,具体方法参考试剂盒说明书。电子荧光显微镜下拍照,PS软件测量细胞面积及JC-1荧光面积,实验共进行3次。
1.9 实时荧光定量PCR
采用QRT-PCR检测H9c2细胞Bax、Bcl-2、caspase-3和GLO1的mRNA表达水平。将GLP-1R-/-和WT组H9c2细胞分别接种于6孔板(1×105个/孔)24 h,饥饿处理16 h后再用0 μmol/L、50 μmol/L MGO处理细胞24 h。Trizol试剂提取总RNA。逆转录试剂盒(RT-PCR)将RNA逆转录成cDNA。Bax、Bcl-2、caspase-3 、GLO1和GAPDH引物序列如下:Bax,forward 5’-GATCAGCTCGGGCACTTTA-3’:reverse 5’-TGTTTGCTGATGGCAACTTC-3’;BCl-2,forward 5’-CCGGGAGAACAGGGTATGATAA-3’;reverse 5’-CCCACTCGTAGCCCCTCTG-3’;caspase-3,forward 5’-AACGGACCTGTGGACCTGAA-3’;reverse 5’-TCAATACCGCAGTCCAGCTCT-3’;GLO1,forward 5’-GAAGCCTGATGATGGGAAAA-3’;reverse 5’-TCTCAGCATCTCGAATCACG-3’;GAPDH,forward 5’-GACATGCCGCCTGGAGAAAC-3’;reverse 5’-AGCCCAGGATGCCCTTTAGT-3’。Bax、Bcl-2、caspase-3 和GLO1的表达水平标准化为GAPDH。Bax、Bcl-2、caspase-3 和GLO1的相对表达量用2-△△Cq法计算。实验共进行3次。
1.10 统计学分析
采用 SPSS 21.0统计软件进行统计分析。统计数据以均数±标准差表示,进行正态分布和方差齐性检验,多组间样本均数比较采用单因素方差分析,P<0.05为差异有统计学意义。
2 结果与分析
2.1 CRISPR/Cas9产生GLP-1R-/- H9c2细胞
将4条gRNA同时转染,最终#B9细胞株被证实为阳性克隆。菌落PCR鉴定阳性转化子,F1/R1显示#B9较野生型细胞缺失295 bp,F1/R2显示#B9较野生型细胞缺失601 bp(表2和图2)。菌落鉴定得到的阳性克隆安排质粒小提并将小提质粒送测序公司进行测序验证,软件比对测序结果分析显示,#B9细胞株等位基因1有296 bp缺失、2 bp发生移码,等位基因2有295 bp缺失、3 bp发生移码,表明#B9细胞株属于杂合突变(图3)。免疫印迹结果显示,WT组细胞表达GLP-1R蛋白,而GLP-1R-/-组细胞GLP-1R蛋白表达缺失(图4)。结果表明,WT H9c2心肌细胞表达GLP-1R蛋白,GLP-1R表达缺失的GLP-1R-/-H9c2细胞构建成功。

表2 PCR鉴定引物

注:M表示相对分子质量标志物,1表示#B9细胞株鉴定引物F1/R1,2表示WT H9c2细胞株鉴定引物F1/R1,3表示#B9细胞株鉴定引物F1/R2,4表示WT H9c2细胞株鉴定引物F1/R2。
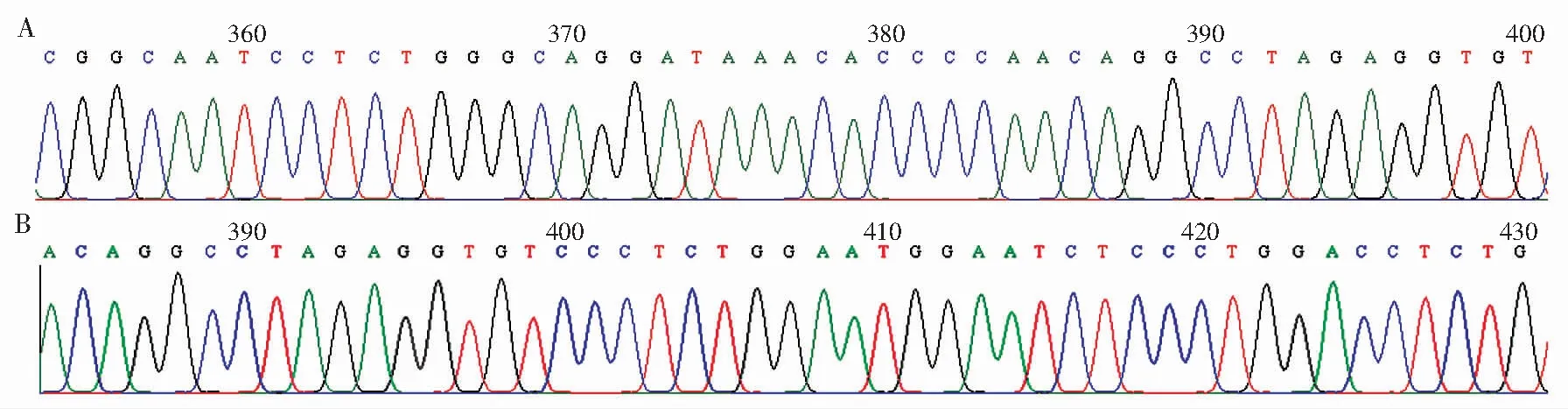
注:A表示等位基因1,B表示等位基因2。
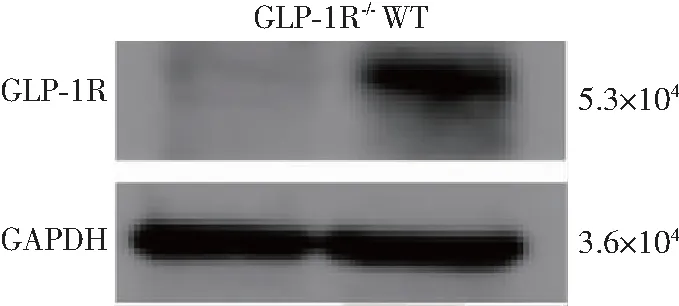
图4 H9c2细胞GLP-1R蛋白的表达
2.2 GLP-1R-/- H9c2细胞形态
构建GLP-1R-/-H9c2细胞模型后,笔者通过细胞染色观察GLP-1R-/-H9c2细胞形态和细胞核改变。图5A显示WT H9c2细胞组呈现舒张状态形态较为多样;GLP-1R-/-H9c2细胞伪足收缩,形态更趋向于圆形。图5B PS软件测量细胞表面积分析表明,GLP-1R-/-组细胞表面积明显小于WT组。

注:Merge,细胞骨架及细胞核染色合成图;Hoechst,细胞核染色;Actin-Tracker,细胞骨架染色。**表示与WT组对比,P<0.01(n=36)。
2.3 GLP-1R抑制MGO诱导的H9c2凋亡
用CCK-8试剂检测MGO对H9c2细胞活力的影响。如图6A显示,MGO浓度为0~40 μmol/L时对WT组H9c2细胞未显示出细胞毒性作用,然而GLP-1R-/-H9c2细胞活力却显著下降。MGO以剂量依赖的方式降低H9c2心肌细胞的存活率,并且MGO的细胞毒性在GLP-1R-/-H9c2中表现更为突出。此外,GLP-1R-/-组较WT组H9c2细胞在经过隔夜饥饿处理后,细胞活力显著下降。用60 μmol/L的MGO干预24 h后,WT组H9c2细胞存活率约为45%,而GLP-1R-/-H9c2细胞存活率约为14%。40 μmol/L的MGO干预24 h后,WT组H9c2细胞存活率约为92%,而GLP-1R-/-H9c2细胞存活率约为60%,因此,选用50 μmol/L浓度进行后续实验。如图6B所示,两组细胞在同等浓度MGO(50 μmol/L)干预下,细胞活力呈现时间依赖性下降,且GLP-1R-/-H9c2细胞对MGO的耐受性更差。

注:*表示与0 μmol/L组内对比,P<0.05;**表示与0 μmol/L组内对比,P<0.01;##表示与相同干预浓度组间对比,P<0.01(n=3)。
2.4 MGO造成线粒体功能障碍
用JC-1检测MMP以评价MGO对H9c2细胞线粒体的影响。发射波长比为590 nm:530 nm。如图7A所示,H9c2细胞在MGO (50 μmol/L)孵育1 h后绿色荧光显著增强,线粒体功能障碍,且GLP-1R-/-组较WT组H9c2细胞更为显著。PS软件测定红绿荧光比值,如图7B,MMP在MGO(50 μmol/L)孵育1 h后开始显著降低,表明MMP去极化,提示线粒体功能障碍,MMP的这种下降可能进一步触发细胞凋亡(与对照组相比P<0.01)。

注:Merge,红色荧光及绿色荧光合成图;Green,绿色荧光;Red,红色荧光。##表示与50 μmol/L WT组对比,P<0.01(n=3)。
2.5 GLP-1R可阻止MGO诱导的H9c2细胞线粒体凋亡通路
如图8A、8B和8C所示,在50 μmol/L MGO干预24 h后,两组细胞的Bax、caspase-3 mRNA表达水平相比于各对应空白组有显著升高(P<0.05)。此外,GLP-1R-/-组的Bcl-2 mRNA表达量显著降低(P<0.01)。其中,两组空白对照组Bax、caspase-3和Bcl-2 mRNA表达无显著差异。但MGO干预24 h后,GLP-1R-/-组Bax mRNA表达水平显著高于WT组,Bcl-2 mRNA表达水平显著低于WT组。如图8D所示,两组细胞在50 μmol/L MGO干预24 h后GLO1 mRNA表达水平相比于各对应的空白组显著降低(P<0.05),与WT组相比,GLP-1R-/-组GLO1 mRNA表达水平降低更明显。
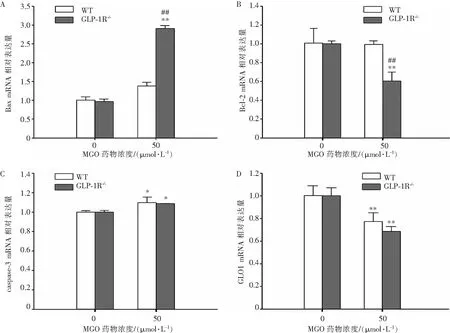
注:*表示与0 μmol/L组内对比,P<0.05;**表示与0 μmol/L组内对比,P<0.01;##表示与相同干预浓度组间对比,P<0.01(n=3)。
3 讨论
在本研究中,首次发现GLP-1R蛋白表达缺失会影响H9c2心肌细胞形态。揭示了GLP-1R在抑制MGO诱导的H9c2心肌细胞凋亡的潜在机制。GLP-1R具抗凋亡作用,包括通过抑制线粒体凋亡通路激活和解毒系统GLO1表达下调,发挥抗凋亡作用。
DCM是一种特异性心肌病,指糖尿病患者在无其他心血管疾病的情况下发生的心功能障碍,而氧化应激的增加与炎症途径的激活会导致心肌细胞功能障碍[2]。MGO能修饰细胞蛋白质和核酸,使细胞功能发生改变[25]。在心肌细胞中,MGO使硫氧还蛋白发生糖基化,使其抗凋亡功能受到抑制,进而加重了心肌细胞的缺血再灌注损伤[26]。MGO的积累增加氧化应激和诱导线粒体功能障碍,促进了心肌细胞凋亡[10]。本研究通过CCK-8检测MGO诱导的H9c2心肌细胞活力,MGO使H9c2心肌细胞活力显著下降,这一现象与Nuamnaichati等[10]用1 mmol/L MGO干预H9c2细胞24 h后通过MTT法检测细胞活力显著下降的研究结果一致,证明MGO在DCM发展中扮演着重要角色。此外,Nuamnaichati等[10]的研究还证实MGO使H9c2细胞活性氧产生显著增加和线粒体功能障碍,从而诱导细胞凋亡,GLP-1R激动剂exendin-4通过cAMP/Epac/PI3K/Akt信号通路在H9c2细胞中表现出抗氧化和抗凋亡作用。
GLP-1R信号通路的激活通过凋亡减轻、氧化应激减弱和能量代谢改善等发挥心肌保护作用[18-21]。然而,这些基于细胞水平的GLP-1R依赖的心肌细胞保护通路的研究均忽略了人体内GLP-1短暂的半衰期。另一方面,GLP-1R的表达在不同物种和不同个体之间存在差异,并且检测方法的敏感性和特异性仍有待提高,因此GLP-1R在心脏的精确定位存在困难[12]。本实验通过蛋白质免疫印迹证实了WT H9c2细胞表达GLP-1R蛋白。
CRISPR/Cas9是依赖RNA识别DNA的基因编辑工程技术,通过改变与Cas9相关的gRNA的序列产生DNA位点断裂[27]。多个不同的gRNA和Cas9进行编程产生多次中断,剪掉染色体的大片段删除目的基因。本研究以GLP-1R基因为研究对象,通过CRISPR/Cas9技术实现了GLP-1R-/-H9c2细胞系构建。本实验首次基于CRISPR/Cas9基因编辑技术构建GLP-1R-/-H9c2细胞系,发现GLP-1R-/-组相比于WT组,H9c2心肌细胞面积显著减小,并且细胞形态呈圆形。DCM患者心脏脂肪酸/葡萄糖氧化代谢失衡,而GLP-1R可介导这种能量代谢失衡恢复。GLP-1处理离体心肌细胞后,心肌细胞的葡萄糖利用率显著升高[28]。本研究表明,对H9c2细胞进行1%低血清隔夜饥饿处理后,GLP-1R缺乏,H9c2细胞凋亡率较WT显著升高。笔者推测GLP-1R-/-H9c2细胞显著减小,与细胞能量代谢受损相关。
本研究通过梯度MGO干预GLP-1R-/-组及WT组H9c2细胞发现,同等浓度MGO干预24 h,GLP-1R-/-组的细胞活力显著低于WT组。当MGO的浓度为50 μmol/L时,能使细胞活力显著下降。在MGO干预后,两组细胞的活力呈现时间依赖性下降,并且在相同时间同等浓度干预下,GLP-1R-/-组细胞活力较WT组低。与神经细胞相比,心肌细胞在更低浓度MGO干预下即出现了明显的损伤,表明心肌细胞对于MGO有着更低的耐受力。GLP-1R-/-组H9c2细胞较WT组H9c2细胞更为脆弱,这也印证了GLP-1R的心肌保护作用。
线粒体途径就是内源性凋亡途径,在应对刺激损伤因素时,Bcl-2被抑制,Bax和Bak被激活,凋亡基因与抗凋亡基因的表达失衡使线粒体膜通透性发生改变,线粒体释放细胞色素c,级联下游含半胱氨酸的caspase造成细胞成分的切割,促使细胞死亡[7]。Bcl-2家族是一组包含了抗凋亡蛋白(Bcl-2、Bcl-xL和Bcl-W)和促凋亡蛋白(Bax、Bak、Bad和Bid)的凋亡调节蛋白[29]。当细胞面临各种应激时抗凋亡蛋白和促凋亡蛋白表达的改变在细胞存活中发挥重要作用。应激条件下,细胞质中的Bax更多锚定至线粒体外膜上并与Bak形成二聚体,最终线粒体外膜上会形成脂质孔,导致细胞色素c从线粒体释放及与凋亡激活因子结合来调节细胞凋亡,同时Bcl-2凋亡抑制作用受阻。艾塞那肽刺激GLP-1R通过Epac减弱了H2O2诱导的活性氧产生、减少凋亡细胞的数量、抑制caspase-3活性和增强抗凋亡蛋白Bcl-2的表达而发挥抗凋亡作用[20]。在本研究中,GLP-1R-/-组H9c2细胞受MGO刺激后MMP较WT组显著下降,线粒体发生严重损伤。此外笔者还观察到MGO打破了Bcl-2与Bax之间的平衡,并且这种失衡在GLP-1R-/-H9c2细胞中更为突出,这些数据印证了GLP-1R通过抑制MGO诱导的线粒体凋亡通路发挥心脏保护作用。caspase-3切割关键的细胞蛋白,导致经历凋亡的细胞发生典型的形态学变化[30]。目前本研究表明,MGO诱导心肌细胞caspase-3表达上调,并且在GLP-1R缺失的心肌细胞中更为显著。
MGO会在细胞和细胞外蛋白质、脂质和DNA上快速生成糖基化加合物,进而改变其生物学功能[11]。MGO对蛋白质的修饰可造成细胞内外蛋白质的功能丧失、交联以及细胞功能紊乱[31]。GLP-1R是经典的7次跨膜受体,MGO干预下GLP-1R对细胞的保护功能可能受到干扰[32-33]。在本实验中,通过梯度MGO干预心肌细胞后,细胞凋亡率表现为显著的浓度依赖性,也证实了MGO可造成显著的细胞功能紊乱,进而促进细胞的凋亡。而在组间对比中发现,GLP-1R缺失心肌细胞呈现更高的凋亡率。另有研究[34]发现,将MGO加入细胞培养基中,MGO定量地被转化为D-乳酸,证实了胞外的MGO进入细胞进行解毒。细胞内的脱毒系统主要是GLO系统[11]。有研究[22]表明,糖尿病患者脂肪组织中GLO1的活性降低,利拉鲁肽提高了GLO1的活性。因此,GLP-1R可能通过改善GLO1的活性及表达来增强对MGO的代谢,进而抑制细胞凋亡。本实验数据证实与空白组比较,MGO干预组GLO1 mRNA表达显著降低。GLO1表达量的下调使MGO的解毒能力受到限制,造成MGO在细胞内的蓄积,MGO进一步通过糖基化作用导致细胞功能紊乱,加速了细胞凋亡。虽然50 μmol/L MGO干预24 h后GLP-1R-/-组及WT组GLO1 mRNA表达无显著差异,但GLP-1R-/-组GLO1 mRNA整体趋势低于WT组。因此,GLP-1R缺失可能通过影响GLO1表达,细胞对MGO解毒能力下降,进而影响细胞的存活率。
总之,本研究成功构建了GLP-1R-/-H9c2细胞系。一方面印证了GLP-1R缺失加剧了MGO诱导的心肌细胞促凋亡信号Bax的表达,下调抗凋亡信号Bcl-2的表达,开启线粒体凋亡通路进一步级联下游切割caspase-3,促进细胞凋亡。另一方面证实GLP-1R缺失会抑制MGO解毒系统关键酶GLO1的表达,进而加剧MGO诱导的氧化应激和凋亡,这些数据均强调了GLP-1R的心脏保护作用。
