中药颗粒剂中不可接受微生物的溯源分析和风险评估
2022-10-12沈振徐晓洁任丽宏冯丹阳孟晓丽丁勃
沈振,徐晓洁,任丽宏,冯丹阳,孟晓丽,丁勃,2
(1.山东省食品药品检验研究院,山东省食品药品安全检测工程技术研究中心,山东 济南 250101;2.山东大学药学院,山东 济南 250012)
中药颗粒剂属于非无菌药品,根据《中国药典》2020年版(四部)通则“1107非无菌药品微生物限度标准”(通则1107)[1],产品中微生物负载需控制在标准限值内,并不得检出大肠埃希菌。中药颗粒剂因其种类繁多的原辅料,独特的生产工艺,易受到来自土壤和水环境的微生物污染,因此,现行标准收载的控制菌种类,对于控制产品质量并不全面,应结合产品微生物负载量、污染微生物特性、药品特性、给药途径、用药人群等方面进行风险评估,将具有潜在危害性的微生物订入产品的微生物限度内控标准或放行标准。
具有潜在危害的微生物即多种国外药典已收载并定义为“不可接受微生物”,通常是指那些已知有明显致病性的,存在足够的数量,通过产品的给药途径可能导致患者不可接受风险的微生物[2]。常见的不可接受微生物包括:肠杆菌科的细菌,如阴沟肠杆菌、肺炎克雷伯菌、粘质沙雷菌、变形杆菌;非发酵革兰阴性菌,如伯克霍尔德菌、嗜麦芽窄食单胞菌、假单胞菌、不动杆菌、鞘氨醇单胞菌等;芽孢杆菌属的细菌,如枯草芽孢杆菌、蜡样芽孢杆菌等[2]。对于非无菌产品,这些不可接受微生物在美国食品药品监督管理局(FDA)发布的药品和医疗产品召回案例中,所占的比例高达70%,其中最常见是伯克霍尔德菌属、假单胞菌属和肠杆菌科细菌[3]。非无菌药品生产企业应对分离到的微生物进行风险评估,确定其是否不可接受,建立微生物测试方法,并制定不低于药典标准的微生物限度放行标准。
本研究源于中药颗粒剂微生物限度检查过程中需氧菌总数偏离正常检验水平的事件,进而分离、纯化和鉴定,确定了主要污染菌。通过生产环节采样,环境微生物的分离、鉴定和核糖体分型分析,溯源污染微生物,并根据污染菌特征,从方法、管理、设备和人员等方面作风险评估并提出应对措施,为生产企业污染微生物的溯源和控制提供依据和方法。
1 材料
1.1 培养基和试剂 胰酪大豆胨液体培养基(批号:20201212)、胰酪大豆胨琼脂培养基(批号:20200507)购自青岛海博生物技术有限公司;pH 7.0氯化钠-蛋白胨缓冲液(1091711)、沙氏葡萄糖琼脂培养基(批号:1090021)、紫红胆盐葡萄糖琼脂培养基(批号:1082281)、麦康凯液体培养基(批号:1087341)、麦康凯琼脂培养基(批号:1081931)购自广东环凯微生物科技有限公司;革兰氏染色试剂(批号:6106099)购自北京陆桥技术股份有限公司;聚山梨酯80(批号:20210107)购自国药集团化学试剂有限公司。
1.2 仪器 VITEK2 Compact型全自动微生物分析系统(法国生物梅里埃公司);RiboPrinter 全自动微生物基因指纹鉴定系统(美国海净纳);DM1000显微镜(德国莱卡公司);GHP-9270隔水式恒温培养箱(上海一恒科学仪器有限公司);IPP250低温培养箱(德国美墨尔特公司);ML4002T/02电子天平(梅特勒-托利多公司)。
1.3 样品 该样品为颗粒剂,13味中药,经煎煮浓缩,浸膏喷雾干燥制粒,与辅料混合干法制粒而得,不含药材原粉。本次检查样品为A、B、C共3个批次。
2 方法与结果
2.1 微生物限度检查 本品为非无菌不含药材原粉的中药颗粒剂,照《中国药典》2020年版(四部)通则1107(四部通则1107)非无菌药品微生物限度标准,应检查需氧菌总数、霉菌和酵母菌总数、大肠埃希菌。
2.2 计数方法适用性试验 称取该样品,以pH 7.0无菌氯化钠-蛋白胨缓冲液作为稀释液,制成1∶10和1∶100供试液,采用平皿法(倾注法),照《中国药典》2020年版(四部)通则1105“非无菌产品微生物限度检查法:微生物计数法”(四部通则1105)进行方法适用性试验,各试验菌回收比值均在0.5~2.0,符合药典要求(见表1)。本品采用计数方法适用性试验确认的方法进行需氧菌总数、霉菌和酵母菌总数的检查(见表2)。
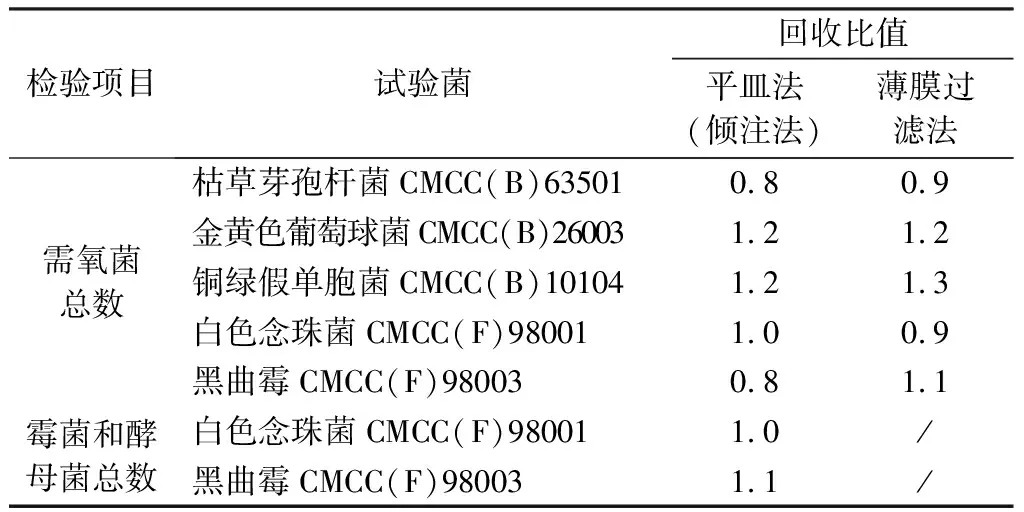
表1 方法适用性试验结果

表2 微生物限度检查结果
2.3 控制菌检查方法适用性试验 取1∶10供试液10 mL,接种至100 mL胰酪大豆胨液体培养基中,照《中国药典》2020年版(四部)通则1106“非无菌产品微生物限度检查法:控制菌检查法1106”(四部通则1106),开展控制菌检查方法适用性试验,在规定的温度和最短时间下培养,可检出大肠埃希菌的反应特征,符合药典要求。本品采用控制菌检查方法适用性试验确认的方法进行大肠埃希菌检查(见表2)。
2.4 样品中污染微生物的分离和鉴定
2.4.1 微生物分离 平皿法(倾注法)琼脂平板上的菌落难以挑取,A批次中需氧菌负载大,取1∶10供试液检查会使菌落计数受到背景杂质和颜色的影响。因此,照“2.2”项下方法制备供试液,取1∶100供试液,采用薄膜过滤法,以pH 7.0无菌氯化钠-蛋白胨缓冲液作为冲洗液,冲洗3次,每次100 mL/膜,滤膜贴在胰酪大豆胨琼脂平板上,33 ℃培养24 h。方法适用性试验照《中国药典》2020年版(四部)通则1105进行,各试验菌回收比值均在0.5~2.0,符合药典要求(见表1)。
2.4.2 菌株纯化 贴有滤膜的胰酪大豆胨琼脂平板上,共计数单菌落28 cfu,根据菌落特征差异,筛选出3个菌落,编号X1、X2和X3。其中,与X1相同菌落特征共26 cfu,X2和X3各1 cfu。用无菌接种环挑取以上3个菌落,划线接种于胰酪大豆胨琼脂培养基上,33 ℃培养18 h。因X1在样品中的生物负载高,超过了需氧菌总数的限度要求,本文将其作为重点研究对象,开展溯源分析。
2.4.3 菌种鉴定 挑取以上3株菌纯培养物,进行革兰氏染色镜检,同时划线接种于麦康凯琼脂和紫红胆盐葡萄糖琼脂培养基上,33 ℃培养48 h。X1、X2和X3均为革兰阴性杆菌,无芽孢,可在麦康凯琼脂平板和紫红胆盐葡萄糖琼脂平板上生长(见表3)。

表3 样品中污染微生物的形态观察
采用VITEK 2 Compact全自动微生物分析系统对3株菌进行生理生化鉴定。X1为阴沟肠杆菌复合菌(Enterobactercloacaecomplex),X2为嗜麦芽窄食单胞菌(Stenotrophomonasmaltophilia),X3为鲁氏不动杆菌(Acinetbacterlwoffii)。
2.5 污染微生物调查和溯源
2.5.1 生产环节采样 经生产现场勘查,发现总混间地面PVC地板有裂缝,且存有积水。根据污染微生物的生物学特性,在了解该品种的生产工艺流程的前提下,围绕总混间,确定了7处采样点:总混间纯化水、总混间地面PVC地板裂缝、总混机内壁、总混间回风口、总混机附近地面、清洗间地面积水处、清洗工具。以含1%聚山梨酯80的胰酪大豆胨液体培养基作为增菌培养基,分别用无菌棉球,每个采样点采样2份,置33 ℃培养7 d,每天观察。
2.5.2 生产环节污染微生物的分离、纯化和鉴定 取生长浑浊的培养液,划线接种在麦康凯琼脂平板和紫红胆盐葡萄糖琼脂平板上,33 ℃培养24~48 h,分离菌落特征存在差异的单菌落,编号Y1、Y2、Y3、Y4、Y5、Y6。分别挑取以上6个单菌落划线在胰酪大豆胨琼脂平板上,33 ℃培养18 h。采用VITEK 2 Compact全自动微生物分析系统对分离得到的纯培养物进行生化鉴定(见表4)。
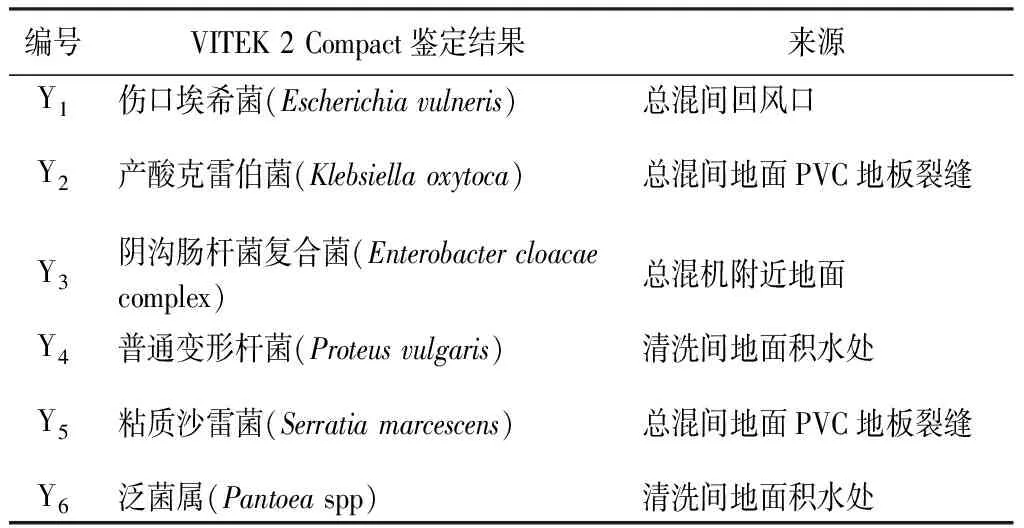
表4 生产环节污染微生物鉴定
2.5.3 同源性分析 挑取VITEK 2 Compact结果一致的X1和Y3两株菌,用RiboPrinter全自动微生物基因指纹鉴定系统进行核糖体分型分析(见图1)。
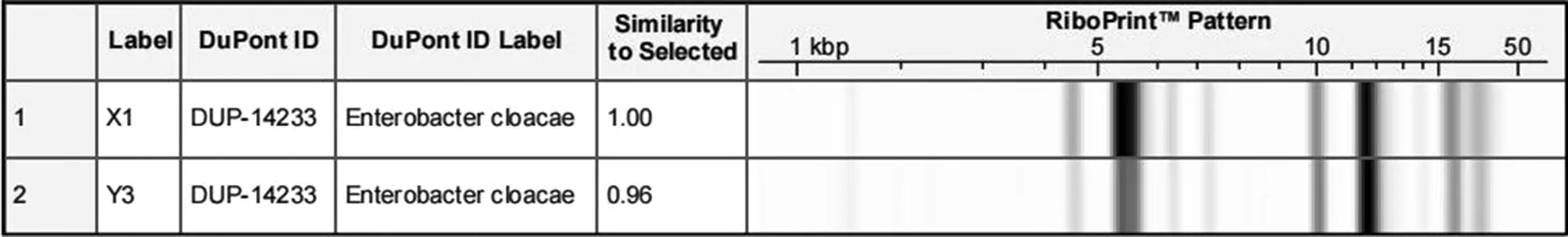
图1 RiboPrinter全自动微生物基因指纹鉴定结果
结果表明,X1和Y3两株菌与RiboPrinter系统库中DUP-14233亲缘关系接近,鉴定为阴沟肠杆菌。而将X1的基因指纹图谱定义为1,Y3的相似度达0.96,结合VITEK 2 Compact,可确认X1和Y3同源。由此说明A批次污染的阴沟肠杆菌主要来源于总混间总混机附近地面。
2.5.4 留样的再检验 抽取与该批次样品同一生产线,生产时间前后共6批次样品,进行微生物限度检查,结果均符合规定,且未检出阴沟肠杆菌和其他生产环节分离的微生物。说明本次微生物污染的事件未覆盖整条生产线,也未波及其他批次药品。
2.5.5 污染微生物风险分析 阴沟肠杆菌分类学上归肠杆菌目肠杆菌科肠杆菌属,是肠道菌群中的一种细菌,同时广泛存在于人和动物的粪便水、泥土、植物中。阴沟肠杆菌是条件致病菌,现已成为医院感染越来越重要的病原菌,其引起的细菌感染性疾病,常累及多个器官,包括皮肤软组织感染、泌尿道感染、呼吸道感染以及败血症等[4-5]。随着临床上青霉素类、头孢菌素、碳青霉烯类抗生素的广泛使用,诱导阴沟肠杆菌能产生超广谱β-内酰胺酶和Amp C酶,耐药情况十分严重,特别对免疫力低下的人群有极大的危害,给临床治疗带来了新的挑战[6-8],同时也给药品生产企业的质量控制加大难度。
虽然调查结果显示本次微生物污染事件未覆盖整条生产线,也未波及其他批次药品,但是,根据产品特点、生产工艺判断,本品容易受到肠杆菌属污染,总混间回风口检出的伤口埃希菌,表明该菌属甚至可以在洁净厂房空气中长期存活,进一步印证了控制阴沟肠杆菌的必要性。因此,建议企业将阴沟肠杆菌作为本品的不可接受微生物进行控制,订入产品的内控标准或放行标准。
3 讨论
3.1 产品中分离出的其他污染菌分析 除阴沟肠杆菌外,本次实验从样品中分离到的其他2株菌,嗜麦芽窄食单胞菌和鲁氏不动杆菌都是医院感染微生物的重要致病菌来源[9-11]。它们主要分布于土壤和水环境中,易在潮湿环境中生存,均属于条件致病菌,当机体抵抗力降低时引发感染。因嗜麦芽窄食单胞菌和鲁氏不动杆菌,在产品中的负载量低,且未能在生产环节分离到,故暂未将其列为不可接受微生物进行控制。但企业应持续关注,积累检测数据,做好趋势分析,一旦发现检出频次或数量的异常波动,应立刻启动风险分析机制,重新评估控制该两种微生物的必要性。
3.2 生产企业的环境控制 药品生产企业应严格按照《药品生产管理规范》2010年版[12]的相关要求进行环境监控,开展浮游菌、沉降菌和尘埃粒子的监测,关注生产环境的异常变化,如地面的腐蚀和损毁、高效过滤器的完整性、车间的温湿度、洁净区的气流变化等。生产环节分离到的微生物从分类学上属于肠杆菌科,说明现有生产环境已被肠杆菌科细菌污染,且适宜于肠杆菌科细菌的生长和繁殖。造成这一微生物污染风险点的可能原因包括:①人员、原料、设备进场未能严格执行相关SOP,造成肠杆菌科细菌由非洁净区向洁净区的转移;②生产后清场不彻底,存在积水,为肠杆菌科细菌的生长和繁殖提供适宜条件;③清场时使用的消毒剂不能彻底地消杀肠杆菌科细菌,甚至引起了该类细菌的抗性。由此,应加强对生产人员和现场QA的培训,包括生物安全、微生物基础理论、生产工艺、设备操作、进出消毒等内容;缩短环境监控周期,增加对高风险区域表面微生物的采集频次;更换或添加不同类型的消毒剂用于人员进出消毒、设备清场清洁和环境的消杀。
3.3 不可接受微生物的风险管理 《美国药典》(USP42)<1115>指出,针对每一种产品都列出不可接受微生物的清单是不现实的。哪些微生物是“不可接受的”取决于药品属性、给药途径、用药人群等。生产企业有责任判断药品中的微生物是否为不可接受。根据ICH Q9[13]和PDA TR67[2]的风险管理流程,不可接受微生物的风险评估包括风险识别和风险分析与评价两个环节。风险识别,即微生物的鉴定和溯源。风险分析与评价,即从给药途径与使用方法、用药人群和药品特性三个方面探讨污染微生物的风险,同时确定产生危害的原因是污染微生物的致病性,还是生物负载量[14]。本次分离到的阴沟肠杆菌,为条件致病菌,生物负载量较高,且药物的使用人群包括老人、儿童和免疫力较低者,属于中等风险水平,故将其列为为本品中等风险的不可接受微生物。而嗜麦芽窄食单胞菌和鲁氏不动杆菌虽是条件致病菌,但生物负载量低,环境中也未检出,可评估为低风险不可接受微生物,暂不进行控制。通过风险评估,生产企业建立不可接受微生物的风险控制方法,设定内控标准,当日常检验结果异常时,应立即启动OOS调查,快速溯源并消除污染源。
3.4 总结 微生物污染风险控制是药品生产企业质量评估的关键环节,也是飞行检查的重要关注点。根据2014~2018年原国家食品药品监督管理总局发布的药品飞行检查数据,在所有缺陷项中,微生物相关缺陷的占比高达36%,且多为高风险缺陷,而涉及中药生产企业的微生物缺陷占到63%[15]。微生物缺陷分布主要集中在方法、管理、设备、原辅料和人员等方面,这些高风险行为则是引发上市产品微生物污染的主要原因。因此,药品微生物污染风险评价与控制应该得到企业的足够重视,特别是对于标准要求之外的潜在污染微生物,要进行充分的研判,必要时增加控制项目,收紧限度要求,尽可能消除其对产品质量的影响。微生物的控制不能仅仅依赖于终产品检验,必须从生产过程控制着手[16-17],持续的微生物质量控制应该贯穿于产品的整个生命周期。企业应培养具有较高微生物鉴定和溯源能力的专业技术人员队伍,建立多元化的微生物鉴定溯源平台,构建环境和产品污染菌库[18],提升微生物污染控制的综合能力。
