T 细胞活化技术的研究进展
2022-10-09张宇煊谢仕廷王鸿鑫叶才果白银山
张宇煊,谢仕廷,王鸿鑫,叶才果,白银山*
(佛山科学技术学院 生命科学与工程学院,广东 佛山 528225)
近年来,随着环境的恶化和人们生活压力的增大,恶性肿瘤的患病率逐年攀升。据统计,到2020 年全世界估计有1 930 万例新肿瘤病例和近1 000 万例肿瘤患者死亡,预计到2040 年,全球肿瘤负担将达到2 840 万例,比2020 年增加47 %[1]。传统的手术、放疗和化疗等肿瘤治疗方法具有一定局限性,很多肿瘤的复发、转移等问题仍无法得到有效的治疗[2]。如化疗的应用前景目前受到肿瘤多药耐药性(Multidrug resistance,MDR)、药物分布的非特异性和系统性毒副作用等缺点限制。迫切需要开发更有效和毒性更低的新型肿瘤治疗方法。最新的纳米载体可以将化疗与基因治疗联合应用,发挥两者的协同作用来改善化疗效果,是一种具有广阔前景的抗癌策略。但是其复杂程度和如何精准控制药物释放时间及部位成为难点和急需解决问题[3]。而随着肿瘤学和免疫学技术的发展,肿瘤免疫治疗的T 细胞活化技术显示出对肿瘤细胞的靶向性和针对性,成为未来肿瘤免疫治疗最有前景的技术之一[4-6]。T 细胞是机体监测细胞突变和防御外界微生物入侵的主力军,分为CD4+和CD8+两大类亚群,两者活化后可分别分化为辅助性T 细胞(helper T cell,Th)和细胞毒性T 细胞(cytotoxic T lymphocyte,CTL),在抗肿瘤免疫中发挥重要作用,介导免疫应答[4-9]。
一些微生物分泌的物质,可以直接高效激活CD4+T 细胞[10-11],被称为超抗原(superantigen,SAg)。这类超抗原在T 细胞活化和在抗肿瘤中显示出了很好的效果[12],但如何合理精准掌控和利用SAg 激活T 细胞,靶向抗肿瘤和减少毒副作用是目前急需解决的问题[13]。双特异性抗体(bispecific antibodies,BsAbs)技术和嵌合型T 细胞(chimeric antigen reportor T cell,CAR-T)技术可以人为地特异性激活CD8+T 细胞,是目前最具前景的肿瘤免疫治疗技术,它们不依赖于T 细胞受体(T cell receptor,TCR)而通过肿瘤抗原依赖方式靶向活化CD8+T 细胞,从而发挥肿瘤杀伤作用[14-16],但也常伴随细胞因子风暴引发的毒副作用[17-19],限制了这些技术临床应用。因此如何高效精准活化T 细胞来靶向杀灭肿瘤并降低副作用成为当前研究的重点。本文重点综述了T 细胞活化技术进展以促进对肿瘤治疗研究和探索。
1 CD4+T 细胞的激活与分化
1.1 超抗原激活CD4+T 细胞
SAg 是来自某些细菌分泌的水溶性蛋白外毒素,可以绕过抗原递呈直接在MHC II 外沟和TCR 的Vβ 区域交叉结合,形成MHC II-SAg-TCR-Vβ 复合物,促使CD4+T 细胞活化和增殖[20],见图1。研究显示每一种SAg 都可以专一地与一种或数种TCR Vβ 亚群结合[21],如葡萄球菌肠毒B(SEB)主要结合人类T 细胞TCR Vβ 3、12、14、15、17 和20 等亚群,小鼠中则和Vβ 1、3、7 和8 等结合[22];葡萄球菌中毒性休克综合征毒素-1(TSST-1)和葡萄球菌表皮剥脱毒素(ET)可以与人TCR Vβ 2 相结合[23]。另外,由于MHC II-SAg 复合体与TCR 的结合性较低,有利于SAg 活化一个T 细胞后即解离,再去结合其他T细胞,导致极低浓度的SAg 就能大量活化CD4+T 细胞,其活化效率高达25 %,甚至40 %[24],而常规抗原递呈只能活化0.01%的T 细胞群体[25]。这种高度的T 细胞活化能引发机体内如毒性休克综合征(TSS)、低反应性脓毒症、自身免疫病等各种疾病的发生[26]。此外,还发现SAg 也能在无蛋白酪氨酸激酶SCR家族LCK 激酶存在的情况下,通过激活异源三聚体鸟嘌呤核苷酸结合G 蛋白11(galpha 11)来激活T细胞。这种方式将SAg 信号与磷脂酰肌醇和蛋白激酶C 信号通路联系起来,为药物开发提供了一个新的方向[27]。SAg 作为最有效的T 细胞激活剂之一,可增强免疫系统的抗肿瘤活性并防止肿瘤生长和转移[28],尽管有不良反应,但许多研究已致力于利用SAg 来增强机体抗肿瘤能力并减少不良反应,用来开发有效的肿瘤免疫疗法。
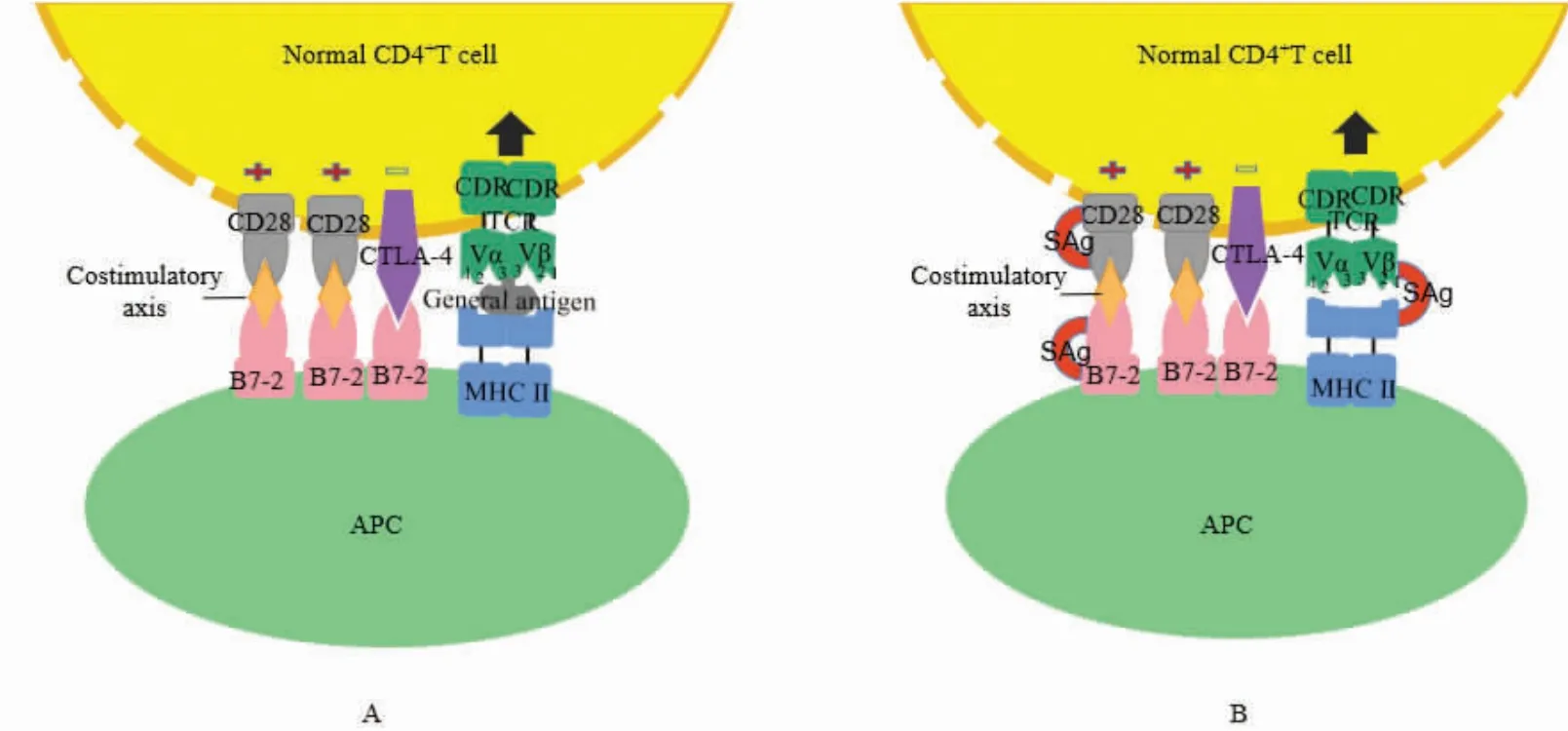
图1 SAg 活化CD4+T 细胞途径示意图
目前,SAg 已被报道的种类和数量有很多,见表1,大部分是外源性T 细胞SAg,介导活化的CD4+T 细胞,具有很强的肿瘤杀伤作用,并已有部分SAg 被应用到医学临床上,如葡萄球菌肠毒素(SEC2)常用于肺癌、胃肠癌、恶性胸腔积液和腹水以及难治性自发性气胸等临床治疗,具有一定的疗效[29-31]。其中,一种从金黄色葡萄球菌变异株的代谢产物中提取的SEC2(高聚金葡素BM828)可显著增加抗肿瘤相关免疫细胞的数量,改善局部免疫反应并延长病人的生存期[32],但存在严重的毒副作用,限制了其广泛应用。HUI 等[33]构建的奥曲肽(SAK)与SEC2 的融合蛋白具有靶向胃癌细胞的功能,此融合蛋白既保留了SEC2 和SAK 的生物活性,又靶向肿瘤细胞表面的受体SST-2,将SAg 抗肿瘤作用限制在肿瘤局部,减少其引起的不良反应,具有较好应用前景,但是否存在潜在脱靶效应还有待进一步考证。SONG等[34]以基因工程的方法融合肿瘤靶向穿膜肽iRGD 和SAg 改构体ST-4,构建出全新的抗肿瘤SAg(ST-4-iRGD),显示出特异的靶向性和良好的肿瘤抑制率,有效提升了实体瘤组织中淋巴细胞的浸润作用,起到更好的免疫效果。这些SAg 的开发与研究显示出了SAg 活化T 细胞的应用前景和优势,它将成为一项重要的T 细胞活化技术,但目前最重要的是精准靶向和降低毒副作用。

表1 T-SAg 的种类
1.2 SAg 通过共刺激信号活化CD4+T 细胞
KRAKASUER 等[44]通过单克隆抗体抑制CD2、CD11a、CD18、CD28、CD44、CD58 和ICAM-1 等,进而抑制了SEB 诱导的T 细胞活化和增殖。阻断ICAM-1 减少了SEB 所诱导的T 细胞产生TNF-α 和IFN-γ,显示SAg 活化CD4+T 细胞同样需要CD11a、CD18/ICAM-1、CD2/CD58、CD28 和CD44 等多种刺激信号。其中,B7-2 和CD28 共受体之间共刺激轴的形成是T 细胞活化的重要刺激信号[45]。KAEMPFER 等[45]研究显示SAg 能直接与B7-2 和CD28 的同源二聚体界面结合,极大地增强了B7-2/CD28 的刺激信号,造成了CD4+T 细胞的过度活化,开启了促炎反应。而NOËL 等[46]发现在小鼠中反复注射SAg 会导致CD4+T 细胞高表达细胞毒T 淋巴细胞相关抗原4(cytotoxic T-lymphocyte-associated protein 4,CTLA-4,也称为CD152),CTLA-4 与CD28 竞争B7 分子配体,结合产生负调控信号,通过减少IL-2 产生和抑制细胞增殖等防止T 细胞过度活化,见图2。CTLA-4 在多种肿瘤中过表达会导致肿瘤生长失控,利用CTLA-4 抑制剂则可促进T 细胞活化和增殖,减少调节性T 细胞(Treg)介导的免疫调节作用[47-48]。

图2 CTLA-4 负调控CD4+T 细胞示意图
1.3 CD4+T 细胞的分化
CD4+T 细胞体内活化后先分化成Th0 细胞,随后在不同因子刺激作用下继续分化为Th1、Th2、Th9、Th17、Th22 和Treg 细胞等亚群,保持动态平衡并相互联系,发挥不同的免疫调节作用[49]。其中,SAg 主要促使Th1 和Th17 细胞亚型增殖,促使分泌大量的IFN-γ 和IL-17A 因子,继而引发“细胞因子风暴”和TSS[44,50]。一些SAg(如SEA)也会诱导Th2 细胞和CD8+T 细胞增加,同时还会抑制Treg 细胞的活性。KRAKAUER 等[51]研究表明SAg 诱导的T 细胞主要向Th1 细胞亚型分化,打破Th1/Th2 平衡,导致促炎因子释放,加重自身免疫性疾病。而XU 等[52]发现SEs 诱导了Foxp3+Treg 细胞增殖和促进IL-35 的产生,有效地抑制了Th1 应答。因此,下调CD4+T 细胞中的IL-35 表达是控制SAg 金黄色葡萄球菌激活Th17 反应的关键条件。
2 CD8+T 细胞的活化
2.1 CD8+T 细胞一般活化
T 细胞中有30 %~35 %是CD8+T 细胞[53],通过TCR 特异性识别APC 细胞膜上的MHC Ⅰ蛋白和抗原肽复合物(pathogenderived peptides-major histocompatibility complx class I,pMHC I),形成以TCRpMHC Ⅰ复合物为中心、周围分布其他共刺激受体的免疫突触,促进CD8+T 细胞活化和增殖,通过分泌颗粒酶、穿孔素或与靶细胞表面的FasL 配体结合,介导杀伤表达Fas 分子的靶细胞[8]。一般情况下,TCR-pMHC Ⅰ-抗原肽是CD8+T 细胞活化的信号,而共刺激受体(CD28-B7、CD40-CD40L、OX40-OX40L 等)能增强其作用[54],见图3,研究发现CD27 在体内促进CD8+T 细胞扩增的能力优于其他的共刺激受体[55]。
尽管CD8+T 细胞的过度激活会导致哮喘、系统性红斑狼疮、移植排斥等多种系统性疾病[56],但适当激活CD8+T 细胞在肿瘤免疫治疗中可发挥重要作用。肿瘤细胞、APC 能够通过高表达程序性死亡配体1(PD-L1)与程序性死亡配体2(PD-L2),与CD8+T 细胞上的程序性死亡受体1(programmed death receptor 1,PD-1)结合,进而抑制CD8+T 细胞活化,导致肿瘤免疫逃逸。而应用PD-L1 抑制剂可阻断PD-1 和PD-L1 的结合[57],重新激活T 细胞来杀伤肿瘤,从而达到抗肿瘤的作用,见图3。另外,TOBIAS等[58]发现CD8+T 细胞中的活化受体CD226 可被肿瘤细胞中的CD155 降解而失去肿瘤杀伤作用,显示出CD226 的表达在肿瘤免疫中具有潜在应用价值。因此,利用CD155 的拮抗剂与PD-1/PD-L1 通路的检查点抑制剂联合使用或许能进一步增强T 细胞杀伤肿瘤能力。
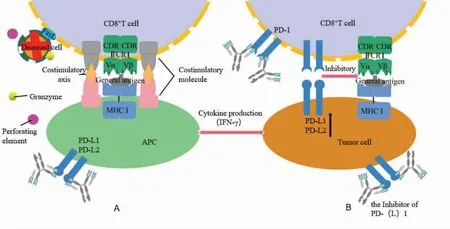
图3 常规肽活化CD8+T 细胞途径示意图
2.2 双特异性抗体激活CD8+T 细胞
BsAbs 是一种人工抗体,拥有两种特异性抗原结合位点,能够识别并结合抗原的两个表位或两种抗原[59]。目前大多数进行临床评估的BsAbs 为双特异性免疫细胞结合分子,主要是T 细胞BsAbs,能够激活机体免疫系统中的细胞免疫而发挥抗肿瘤作用。T 细胞BsAbs 一端靶向肿瘤表面相关抗原(tumorassociated antigen,TAA),另一端则靶向T 细胞[60],通过募集T 细胞到靶标肿瘤细胞附近,进而激活内源性T 细胞,选择性杀伤肿瘤细胞[61]。此技术可以不受组织相容性复合体(MHC)的限制,不依赖于TCR 的表位特异性便能激活T 细胞[62]。大多数T 细胞BsAbs 以T 细胞上的TCR/CD3 复合体为靶点,也有一些以CD2 和CD5 为靶点,但其激活作用都不如靶向CD3[63-65]。而以T 细胞CD28 为靶点的抗CD28抗体曾引起严重的细胞因子释放综合征(CRS),导致危及生命的并发症,一段时间内限制了以CD28 为靶点的BsAbs 发展[66]。T 细胞BsAbs 能够招募不同群体的T 细胞来选择性杀伤肿瘤细胞,激活CD8+T细胞在抗肿瘤方面起主导作用[63,67-68]。
目前,BsAbs 在血液肿瘤治疗方面已获得一定的成效,如Blinatumomab(抗CD19×抗CD3 BiTE)已被美国食品药物管理局(FDA)批准用于治疗复发性或难治性急性淋巴细胞白血病[69],见图4。但在实体瘤方面还有待更加深入的研究,其中如何让BsAb 更加有效的进入肿瘤并在肿瘤微环境中保持其激活及强大的抗肿瘤能力,是BsAb 应用于实体瘤的一个难点。当前已有许多研究致力于突破这一难点,如GARDELL 等[70]经基因工程改造巨噬细胞后,使其分泌出一种BiTE 蛋白,能够诱导T 细胞活化和增殖,有效杀伤肿瘤细胞,阻止其生长,而双重转导巨噬细胞分泌BITE 蛋白和IL-12 能增强这种杀伤力作用。瘤内注射纯化的BiTE 或BiTE 和IL-12 蛋白可以延缓,但不能阻止肿瘤生长,显示了基因工程巨噬细胞(Gem)在实体瘤中的作用。KILIC 等[71]发现双特异性抗EGFR×抗CD3 的化学自组装纳米环(CSANs)能够下调EGFR 信号来减少PD-L1 表达,从而降低肿瘤微环境的免疫抑制作用。治疗的肿瘤有明显的T 细胞浸润和增殖,并伴有肿瘤细胞的凋亡和坏死。而CSANs 可以通过添加药物甲氧苄啶来拆解,从而防止对T 细胞的过度激活,见图4。因此,基于CSANs 的BsAbs 使T 细胞BsAbs 治疗更具可控性,进一步探究其临床效果具有重要的研究意义。

图4 CD8+T 细胞的双抗设计
许多研究表明T 细胞BsAbs 与PD-1/PD-L1 免疫检查点阻断剂[72]、共刺激的CD28 双抗等联合治疗能够增强T 细胞活化或者抗肿瘤作用。SKOKOS 等[66]提出了针对卵巢癌和前列腺癌的两种CD28 双特异性抗体,它们与T 细胞BsAbs 联合使用时,能够显著促进T 细胞活化,增强了T 细胞BsAbs 的抗肿瘤活性,但无使用CD28 激活抗体时引发的全身性毒副作用。目前有许多针对免疫检查点和T 细胞共刺激分子而设计的BsAbs,能增强T 细胞的活化。如FS120 mAb2,一种针对T 细胞共刺激分子CD137和OX40 的BsAb,以FcγR 非依赖性机制同时激活CD4+和CD8+T 细胞;FS118 mAb2 是一种针对LAG-3 和PD-L1 的BsAb,能够克服LAG-3 和PD-L1 介导的T 细胞活化抑制以增强CD8+T 细胞的激活来抗肿瘤[74];MA 等[75]针对一种新的免疫检查点分子(TIGIT)开发了一种新型BsAb,具有PD-L1 和TIGIT双重阻断活性,在体外协同增强激活T 细胞,如图4 所示。因此,针对两种免疫检查点的BsAbs 与T 细胞BsAbs 联合使用时,应能进一步增强T 细胞BsAbs 的抗肿瘤能力。有文献表明,BsAbs 双臂的亲和力也与T 细胞的活化以及抗肿瘤活性相关。如YOON 等[76]构建了新型的T 细胞BsAbs,分别为MG1122-A 和MG1122-B,能够靶向表达间皮素(MSLN)的实体瘤。其中MG1122-B 与MSLN 二价结合,而MG1122-A 是一价结合。实验表明两者都能诱导靶向MSLN 特异性T 细胞活化,但MG1122-B 对肿瘤抗原的亲和力更高,T 细胞活化和肿瘤杀伤的功效更强。另外,ATTACK 是一种具有三价TAA 上皮生长因子受体(EGFR)结合和单价CD3 结合的T 细胞BsAbs,其激活T 细胞的效力是一价EGFR 结合的T细胞BsAbs 的15 倍。然而,T 细胞BsAbs 是否对肿瘤抗原的亲和力越高,其激活T 细胞和杀伤肿瘤的效力就越高,还有待于进一步的研究[69]。
细胞因子风暴是T 细胞BsAbs 治疗的一个常见毒副作用,包括急性呼吸窘迫综合征、肾功能衰竭和肝功能衰竭等[61],如何减少BsAbs 治疗带来的副作用有助于其在临床上的应用。另一个被批准用于临床肿瘤治疗的T 细胞BsAbs 是Catumaxomab(抗EpCAM×抗CD3 全长IgG)[77],但临床显示静脉注射Catumaxomab 诱导了局部细胞强烈的因子释放和CD8+T 细胞介导的肝毒性作用。这种严重不良反应主要是其活性Fc 区与肝脏表达FcγR 的肝内表面的吞噬细胞结合引起的[78],表明T 细胞BsAbs 诱导的毒副作用与其Fc 区密切相关,抑制Fc 介导的效应能够减少非靶向细胞毒副作用并发挥治疗效果。目前利用工程改造的Fc 区减少或消除与FcγR 和补体成分C1q 的结合,这种IgG-like(含Fc 片段)的T 细胞BsAbs 可显著降低毒副作用,但仍能保持与新生儿Fc 受体(FcRns)结合以促进IgG 的体内循环[76]。而non-IgG-like 的T 细胞BsAbs,如Blinatumomab 直接去掉Fc 域,这类BsAbs 具有免疫原性低、副作用少和组织穿透能力强,但其分子量过小,血浆半衰期非常短,患者需要持续静脉输液[79-80]。AFM11,一种四价无Fc 结构的抗CD19×抗CD3 四价串联双抗体(tandem diabodies,TandAbs),其分子量是Blinatumomab 的2 倍,血浆半衰期却延长了10 倍[79,81],这表明在无Fc 结构下,BsAbs 的分子量影响其血浆半衰期-小于约60 kDa 的药物可以通过肾小球滤过迅速消除[65],或许可以通过开发适当分子量结构的BsAbs 来延长循环半衰期,见图4。另外有研究表明,采用低亲和力的抗CD3 臂也能够减少细胞因子的释放,但仍具有较强的抗肿瘤能力。如MALIK-Chaudhry 等[82]采用了低亲和力的抗CD3 臂开发了一种完全人源化抗CD19×抗CD3 的T 细胞BsAbs,称为TNB-486,可以降低人类的免疫原性风险,具有与Blinatumomab 结构同等的细胞毒性,但细胞因子释放减少。另外,有研究发现相同抗肿瘤臂之下,具有高亲和力CD3 结合臂的T 细胞BsAbs 相比低亲和力CD3 结合臂,不仅显示出对脾和淋巴结的定位增加[83],而且在次级淋巴组织中具有较高的分解代谢量,表明较低的CD3 亲和力能够较好地避免向次级淋巴组织的高分布和CD3 介导的血浆清除。因此,开发具有低亲和力CD3 结合臂和高肿瘤抗原结合臂的BsAbs 可能更适合应用于临床。
2.3 嵌合抗原受体T 细胞激活CD8+T 细胞
CAR 是模块化的合成受体,主要由4 个部分组成:1)一个细胞外靶抗原结合结构域(extracellular target antigen-binding domain);2)一个铰链区(hinge region);3)一个跨膜结构域(transmembrane domain);4)一个或多个细胞内信号转导结构域(intracellular signaling domain)[84]。在跨膜结构域中,由于CD3ζ 跨膜结构域介导CAR 二聚化和整入内源性TCR 中,导致其可能促进CAR 介导的T 细胞激活。与具有CD8α 或CD28 跨膜结构域的CAR 相比,具有CD3ζ 跨膜结构域的CAR 稳定性和表达较差。目前,这种模块化结构已从单个CD3ζ 信号结构域扩展到CD3ζ-CD28、CD3ζ-4-1BB 或CD3ζ-CD28-4-1BB 信号结构域来进一步提高CAR 的稳定性和表达效率[15],见图5。
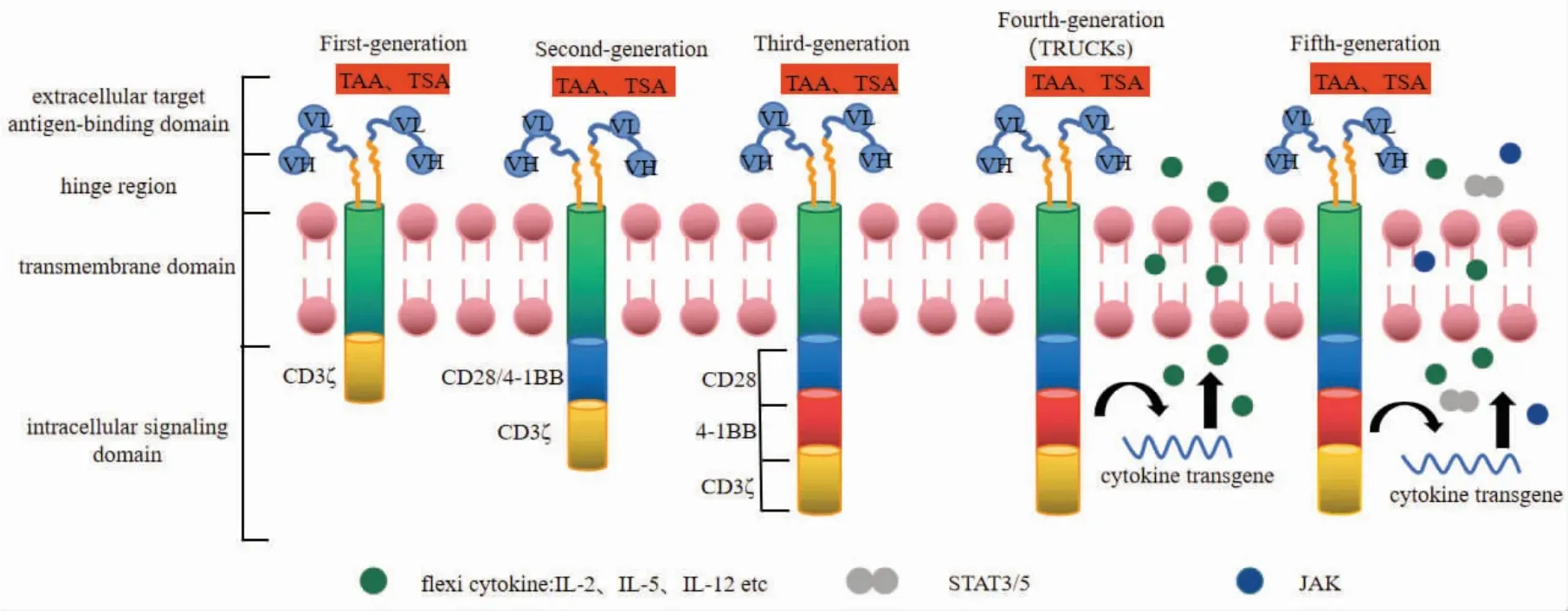
图5 CAR-T 细胞的发展
CAR-T 细胞技术具有肿瘤靶向特异性,活化的T 细胞对肿瘤抗原的杀伤绕过了抗原递呈阶段及MHC I 的限制,导致杀伤活性得以最大化[85]。大多数CAR-T 都是利用单链可变片段(scFv)识别、靶向结合TAA,还有少数能利用融合跨膜信号区的受体配体如anti-VEGFR2,anti-AVB6,anti-Her 3/4 receptor 等瞄定肿瘤[86]。虽然CAR-T 细胞对治疗肿瘤具有很好的疗效,但较高的毒副作用率阻止了CAR-T 细胞疗法成为一线治疗。如何降低CAR-T 细胞技术所带来的毒副作用成为科学家的研究热点之一。研究发现,缓解CAR-T 细胞毒性的一个潜在途径是通过实施“关闭开关”或自杀基因策略。FOSTER 等[87]利用工程化修饰T 细胞使其携带一种名为iC9(诱导性caspase-9)的安全开关,如果出现毒性作用,其就会被激活表达,从而降低因CAR-T 疗法所产生的严重副作用。
全球约90 %的癌症病例是实体瘤,但未满足的临床需求仍十分巨大。CAR-T 疗法攻克实体瘤治疗中的局限,是目前该领域面临的最紧迫的挑战之一。CAR-T 细胞会经历快速和广泛的抑制性DNA 甲基化重编排,导致T 细胞的衰竭而严重限制其在实体瘤中的应用,因此阻止这一过程可能是提高CAR-T细胞在实体瘤中疗效的可行方法[88]。研究发现通过敲除CAR-T 细胞中的DNMT3A 基因[89](编码DNA甲基转移酶3α)或者抑制BET 蛋白[90](破坏T 细胞的乙酰化组蛋白功能)等可以逆转T 细胞衰竭,促使CRA-T 细胞保留活性并进一步增强抗实体瘤的能力。
与血液肿瘤的靶点大多单一且具有特异性不同,目前在实体瘤中发现的肿瘤高表达抗原多为TAA,在正常组织中也有表达,包括CEA、HER2、GPC3、EpCAM 等[91],但肿瘤特异性抗原(TSA)很少,这是CAR-T 细胞在实体瘤中应用困难的另一个原因。目前科学家们主要是通过对CAR-T 细胞本身进行改造[92]、利用外界条件[93]或通过靶向两种或者多种不同TAA 的CAR-T 疗法[94]等来提高CAR-T 细胞对肿瘤细胞的特异性识别,有助于CAR-T 细胞在实体瘤中的应用。
与此同时,CAR-T 细胞的转运和浸润同样对CAR-T 细胞治疗实体瘤带来挑战。研究发现趋化因子是影响T 细胞浸润的重要分子,LI 等[95]首次将趋化因子受体CXCR5 修饰到靶向EGFR 的CAR-T细胞表面,可以定向迁移和渗透至肿瘤病灶处,并显着清除了肿瘤,同时极大减轻了潜在的肿瘤外毒性。另外,肿瘤微环境(TME)中的免疫抑制作用也是影响CAR-T 在实体瘤中发挥效力的关键因素之一。研究发现通过修饰CAR-T 细胞使其过度表达促进炎症的细胞因子(如IL-12、IL-15 和IL-18)[96]、阻断TGF β 信号(一种重要的免疫抑制途径)[97]或使用基因沉默、PD-1 开关受体和与PD-1 抑制剂联用等手段均可以提高CAR-T 细胞的抗实体瘤效果。
3 SAg 激活CD8+T 细胞
SAg 不仅能够激活CD4+T 细胞,还能激活CD8+T 细胞,介导细胞免疫[98]。MEILLEUR 等[99]发现SAg 能活化和增殖已存在的记忆CD8+T 细胞,且活化的CD8+T 细胞仍具有效应记忆表型,能够产生IFN-γ 并在体内外破坏靶细胞。合理利用SAg 高效激活CD8+T 细胞成为肿瘤免疫治疗的另一途径。实验发现,应用超抗原金葡素C 型(SEC)联合食管癌TSA 构建肿瘤疫苗可诱导PBMC 活化增殖,产生CD8+T 细胞,进而靶向肿瘤细胞,起到特异性杀伤作用[100]。也有研究表明,用SEC1 处理PBMCs 后,CD8+T 细胞的水平增加了,能够诱导肿瘤细胞和被病毒感染的细胞死亡[101]。因此,合利用SAg 激活CD8+T 细胞来定向杀伤肿瘤细胞具有重要的研究意义。
4 总结与展望
T 细胞活化是发挥免疫应答的关键步骤,通过SAg、BsAbs 和CAR-T 等技术能够有效激活T 细胞,建立细胞和体液免疫应答。深入研究T 细胞的活化机制,有助于在肿瘤免疫治疗中提供新思路和方法,促进免疫医学和肿瘤治疗的发展。
然而,尽管SAg、BsAbs 和CAR-T 等技术治疗肿瘤后的缓解率很高,但达到持续缓解仍是不小的挑战。在继续强化其对肿瘤杀伤作用的同时,还应努力探索应对毒副作用等的新策略和潜在的解决方案,为未来更有效和更安全的治疗提供一条道路。
