D-阿洛酮糖 3-差向异构酶在枯草芽孢杆菌中的表达
2022-10-05胡梦莹李梦丽江波张涛
胡梦莹,李梦丽,江波,张涛
(江南大学,食品科学与技术国家重点实验室,江苏 无锡,214122)
国际稀有糖协会将自然界中很少存在的单糖和糖醇定义为稀有糖,D-阿洛酮糖就是其中的一种,它是D-果糖在C-3位上的差向异构体[1],早在2014年就被美国食品药品监督管理局列为一般安全,允许将其添加在食品、膳食补充剂以及医药制剂中[2]。D-阿洛酮糖具有许多有益人体健康的特殊功能[3],例如摄入适量的D-阿洛酮糖可以降低血糖血脂水平[4]、减少脂肪堆积[5]、清除活性氧[6]的活性等。且当人体摄入D-阿洛酮糖后,生成的热量较低,仅为同等质量蔗糖的0.3%[7],因此被美国食品导航网评为最具潜力的蔗糖替代品。
目前D-阿洛酮糖的生产主要依赖于酶法生产,这主要是因为其在自然界中含量稀少,直接提取不符合经济效益,而化学合成法会产生副产物,增加生产成本[8]。而酶法生产中最关键的酶是D-阿洛酮糖 3-差向异构酶(D-psicose 3-epimerase,DPE),它可以将D-果糖转化为D-阿洛酮糖。野生型菌株的产酶量远远低于预期,无法应用于工业生产中,因此,有必要构建合适的表达载体并在异源生物中表达。枯草芽孢杆菌是革兰氏阳性菌[9],具有较为清晰的研究背景[10],发酵周期短、分泌能力强且是食品级安全菌株,是一个较好的表达宿主[11]。
本研究中将ClostridiumscindensATCC 35704来源的DPE,在枯草芽孢杆菌(Bacillussubtilis)WB800中进行异源表达,通过筛选单启动子/串联启动子,优化核糖体结合位点序列,从而提高DPE的表达量,为DPE生产应用于食品行业提供实验数据支持。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
菌株大肠杆菌(Escherichiacoli)DH5α、B.subtilisWB800为实验室保藏;穿梭质粒pP43 NMK由实验室保存;质粒PUB-dpe由实验室前期构建而成[12]。
1.1.2 试剂
Phanta Max Super-Fidelity DAN Polymerase、ClonExpress Ⅱ One Step Cloning Kit、质粒快速抽提试剂盒、DNA 凝胶回收试剂盒,南京诺唯赞生物科技有限公司;超级感受态细胞制备试剂盒、细菌DNA基因组提取试剂盒、4S Green Plus无毒核酸染料、琼脂糖、50×TAE 缓冲液,上海生工生物工程有限公司;DNA Marker、10×Loading Buffer,TaKaRa(大连生物有限公司);PAGE凝胶快速制备试剂盒、蛋白质marker,5×蛋白上样缓冲液,上海雅酶生物医药科技有限公司;葡萄糖、酵母提取物、蛋白胨及其他常用试剂,国药集团。
1.1.3 培养基
LB培养基(g/L):酵母提取物5,胰蛋白胨10,NaCl 10,121 ℃高压灭菌20 min(固体中需额外添加20 g/L琼脂)。
发酵培养基(g/L):葡萄糖15,蛋白胨10,酵母浸粉20,NaCl 8,Na2HPO4·12H2O 1,MgSO4·7H2O 1,调节pH至7.0,115 ℃高压灭菌30 min。
1.2 实验方法
1.2.1 引物的设计
本文所用的引物如表1所示,由苏州金唯智生物科技有限公司所合成。

表1 本研究中涉及的所有引物
1.2.2 pP43 NMK-promoter-DPE单启动子表达质粒的构建及克隆
利用不同启动子的引物对Promoter-F/R(表1),以B.subtilis168基因组为模板,通过PCR技术扩增得到启动子PamyE、PaprE、PgsiB、Phag、PnprE、P43、PsigX、PylbP片段,以实验室保存的质粒PMA5为模板,通过PCR技术扩增得到启动子PHpaII片段。以实验室保存的质粒PUB-DPE为模板,利用不同的引物对DPE(prometer)-F/DPE-R,通过PCR技术扩增得到DPE片段。其中引物Promoter-R与引物DPE(promoter)-F有15~20 bp的同源臂,利用重叠PCR可将Promoter片段与DPE片段进行融合,形成新片段Promoter-DPE。产物经琼脂糖凝胶电泳和胶回收,在-20 ℃保存。以实验室保存质粒pP43 NMK为模板,利用引物对P-F/R,去除原有启动子序列,进行PCR线性化扩增。其中引物Promoter-F、DPE-R分别与引物P-R、P-F有15~20 bp的同源臂,通过ClonExpress Ⅱ One Step Cloning Kit试剂盒,将Promoter-DPE片段和线性化pP43 NMK片段环化后构建含不同单启动子的载体pP43 NMK-promoter-DPE。
通过超级感受态细胞制备试剂盒制备E.coliDH5α感受态细胞,并按照说明书方法将pP43 NMK-promoter-DPE转化至E.coliDH5α感受态细胞,将重组细胞涂布在LB 氨苄(100 mg/L)抗性固体平板上,37 ℃过夜培养,挑取转化子,筛选验证测序。将测序验证正确的质粒pP43 NMK-promoter-DPE转至B.subtilisWB800感受态细胞,将重组细胞涂布在LB卡那(50 mg/L)抗性固体平板,37 ℃过夜培养,挑取转化子,测序正确后即得到重组菌株B.subtilisWB800/pP43 NMK-promoter-DPE。
1.2.3 pP43 NMK-promoter1-promoter2-DPE串联启动子表达质粒的构建及克隆
以1.2.2中构建的含有不同单启动子pP43 NMK-promoter-dpe质粒为模板,利用引物对P-F、Promoter1(promoter2)-R,通过PCR技术扩增pP43 NMK-Promoter1片段,利用引物对(promoter1)Promoter2-F、DPE-R,通过PCR技术扩增Promoter2-DPE片段,其中引物P-F、Promoter1(promoter2)-R分别和引物DPE-R、(promoter1)Promoter2-F有15~20 bp的同源臂,通过ClonExpress Ⅱ One Step Cloning Kit试剂盒,将pP43 NMK-Promoter1片段和Promoter2-DPE片段环化后构建含不同串联启动子的载体pP43 NMK-promoter1-promoter2-DPE。
按照1.2.2中的转化方法,最后得到重组菌株B.subtilisWB800/pP43 NMK-promoter1-promoter2-DPE。
1.2.4 RBS序列的优化
文献[13]报道绝大多数细菌中有着通用的SD序列(shine-dalgarno sequence,SD)GGAGG。本文中待优化的核糖体结合位点(ribosome binding site, RBS)同样具有该SD,以此作为RBS序列的种子区域,突变前两个与后两个碱基。报道中的RBS序列里A、G碱基出现频率较高[14-16],因此将前后两个碱基在A/G中随机出现,得到包括对照RBS序列在内的16种RBS序列。
1.2.5B.subtilisWB800重组菌株的发酵培养与表达
用接种环在LB卡那抗性平板上划线,37 ℃培养12 h,挑取单菌落接种至LB培养基中,37 ℃,200 r/min摇床培养12 h。将活化后的种子液按3%的接种量接种至发酵培养基中,37 ℃,200 r/min摇床培养,定时取样,检测菌体生长OD600值和DPE酶活力。
1.2.6 酶活力的检测
吸取1 mL发酵液,4 000 r/min离心5 min,弃上清液,保留菌体沉淀。用50 mmol/L,pH 7.5的Tris/HCl缓冲液反复吹洗菌体沉淀后,将其稀释至合适浓度。取一定量稀释后的发酵液,加入到终体积为1 mL 的果糖底物溶液中(50 mmol/L,pH 7.5的Tris/HCl 缓冲液配制,质量浓度100 g/L),55 ℃,反应10 min后,高温煮沸灭酶。将样品于12 000 r/min离心5 min,上清液过0.22 μm 滤膜,制样,利用HPLC检测分析。
其中HPLC的检测条件为:Waters e2695高效液相,流动相为超纯水(添加50 mg/L EDTA钙盐,超声15 min),流速0.4 mL/min;Sugar pak-1钙型色谱柱,柱温85 ℃;示差折光检测器,检测器温度30 ℃。
酶活力定义:在55 ℃,pH 7.5的条件下,单位时间(min)生成1 μmolD-阿洛酮糖所需要的酶量为1个酶活力单位(U)。
2 结果与分析
2.1 不同启动子重组菌株的构建
利用表1中的引物,对各个启动子进行PCR扩增,启动子片段带有相应的上下游同源臂序列,其PCR产物电泳结果如图1 所示,可知启动子片段大小与预期一致。当单启动子重组菌株B.subtilisWB800/pP43 NMK-promoter-DPE构建完成后,用质粒快速抽提试剂盒抽取该菌株质粒,以其为模板,按照1.2.3中的方法,构建串联启动子重组菌株pP43 NMK-promoter1-promoter2-DPE,其中pP43 NMK-Promoter1片段和Promoter2-DPE片段PCR产物电泳结果见图1,其大小与预期一致。待转化子测序无误后,即可获得重组菌株B.subtilisWB800/pP43 NMK-promoter-DPE和pP43 NMK-promoter1-promoter2-DPE。
a-Promoter片段PCR扩增;b-Promoter2-DPE片段PCR扩增;c-pP43 NMK-Promoter1片段PCR扩增
2.2 单启动子对重组菌株表达水平的影响
靶基因的有效转录是蛋白表达的第一步,启动子在基因组中是重要的调控元件,能够决定基因表达的时间和强度[17]。为了进一步提高DPE的表达效果,构建了9种启动子介导的DPE重组表达菌株,并对其进行发酵产酶,其最大酶活力如图2所示。本文选用的启动子均为枯草芽孢杆菌中表达效果较好的启动子,由图2和图3可知,在发酵产DPE时,不同启动子介导的DPE表达效果差异较大,但启动子对菌体的生长情况影响较小,其酶活力最高时OD600值差异较小。其中最优单启动子为Phag,相应重组菌株酶活力高达19.62 U/mL,OD600值为12.03,其次是启动子PylbP和P43,对应的酶活力分别为16.62 U/mL和15.05 U/mL,OD600值分别是11.87和11.31。Phag由σD识别,在枯草芽孢杆菌中σD涉及鞭毛基因、运动性基因和自溶素基因及其调节因子的表达。在所选启动子中,PsigX是P43强度的3.03倍[18],但在DPE的表达中,效果较差。这些结果表明启动子和目的基因之间存在着一定的适配性,强度大的启动子表达效果不一定好,需要进行不断的筛选,找出最适启动子。
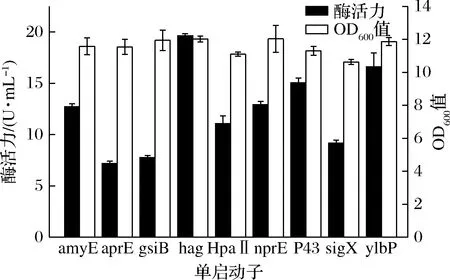
图2 不同单启动子重组菌株酶活力及OD600值
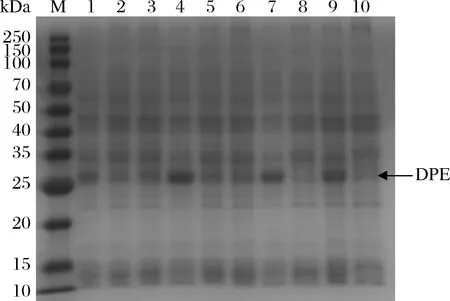
M-250 kDa 蛋白 Marker;1-amyE;2-aprE;3-gsiB;4-hag;5-HpaⅡ;6-nprE;7-P43;8-sigX;9-ylbP;10-对照
2.3 串联启动子对重组菌株表达水平的影响
选取启动活性较高的3个启动子Phag、PylbP和P43进行下一步研究。将这3个启动子进行两两/重复串联,通过构建串联启动子介导的DPE重组表达菌株,研究串联启动子对DPE表达的影响,同时交换被串联的两个启动子顺序,研究启动子的位置对DPE表达的影响,其最大酶活力及SDS-PAGE分析分别如图4与图5所示。大部分启动子经过串联以后,酶活力反而有所降低,推测是因为所筛选的单启动子均为启动强度较强的启动子,当它们被串联时,会彼此形成干扰或竞争,反而使表达水平有所降低[19]。少部分启动子经过串联以后,酶活力有了提升,如PylbP-ylbP,但其最大酶活力依然低于单启动子Phag。同时,改变被串联的两个启动子的顺序,酶活力也随之有了较明显的改变,这说明启动子内部核心序列的位置,对目的基因的表达影响较大,通常而言,与目的基因相邻的启动子,其强度对串联启动子的转录活性影响更大[20]。此外,吴凤依[19]构建了串联启动子PP43-hag介导的重组菌株表达脂肪酶A,其表达效果最好,但对于本文的DPE而言,PP43-hag介导的重组菌株表达效果反而较差,这同样证明启动子和目的基因之间存在着一定的适配性。另外,与单启动子介导的重组菌株相比,串联启动子介导的重组菌株在酶活力最高时,其整体OD600值的范围要略低一些,推测是因为双启动子介导时,对重组菌株的生长造成了一定的胁迫,这也是串联启动子介导的重组菌整体酶活力较低的一个原因。
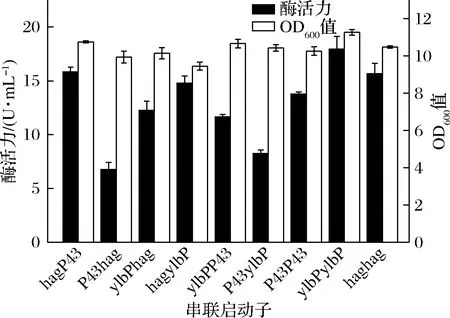
图4 不同串联启动子重组菌株酶活力及OD600值
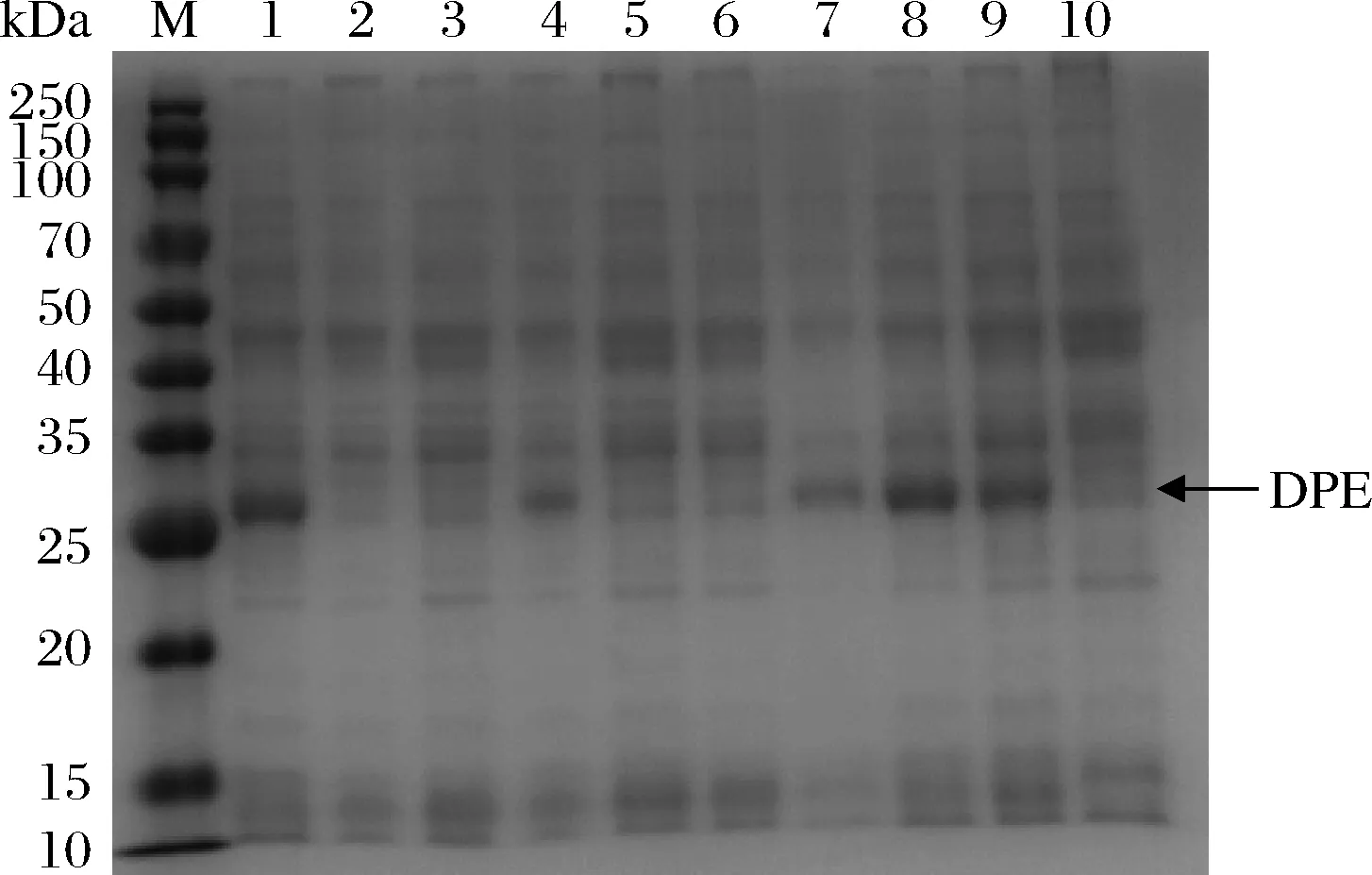
M-250 kDa 蛋白 Marker;1-hagP43;2-P43 hag;3-ylbPhag;4-hagylbP;5-ylbPP43;6-P43ylbP;7-P43P43;8-ylbPylbP;9-haghag;10-对照
以上结果同样也表明,将两个启动活性较高的启动子串联,不一定能够继续增强串联启动子的活性。
2.4 RBS序列对重组菌株表达水平的影响
RBS序列即核糖体结合位点,决定着翻译水平的高低,是蛋白表达的关键区域[21]。它位于起始密码子AUG上游约8~13核苷酸处,是一段富含嘌呤的非翻译区,可以与核糖体16S rRNA互补配对并与之结合,从而控制翻译的起始。经过筛选,发现单启动子Phag介导的重组菌株在表达DPE时效果最好,因此以启动子Phag的RBS序列为对象,GGAGG序列为RBS种子区域,对其前后两个共计4个碱基进行随机A/G突变,旨在提高目的蛋白的表达。RBS序列中具体突变序列如图6所示,突变后的重组表达菌株酶活力及OD600值见图7。RBS序列突变后,大多数重组菌株的酶活力得到了显著提升,其中重组菌株B.subtilisWB800/pP43 NMK-hag-R4-dpe的酶活力最高,为25.39 U/mL,OD600值为11.65,较突变前酶活力提高了29.4%。同时,在各突变菌株酶活力最大时,其OD600值相差不大,说明突变对菌株的生长情况影响较小。
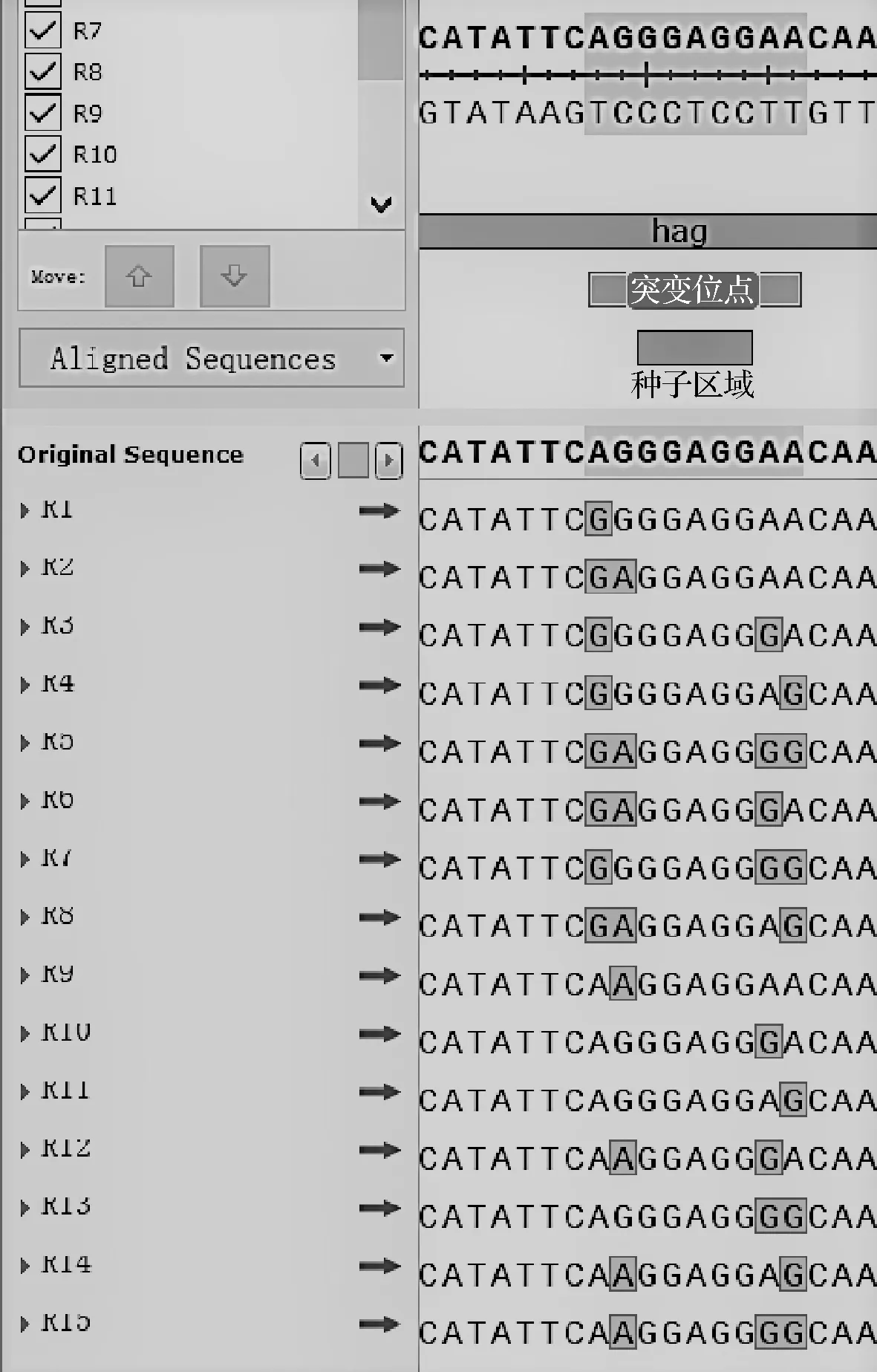
图6 RBS序列不同突变位点
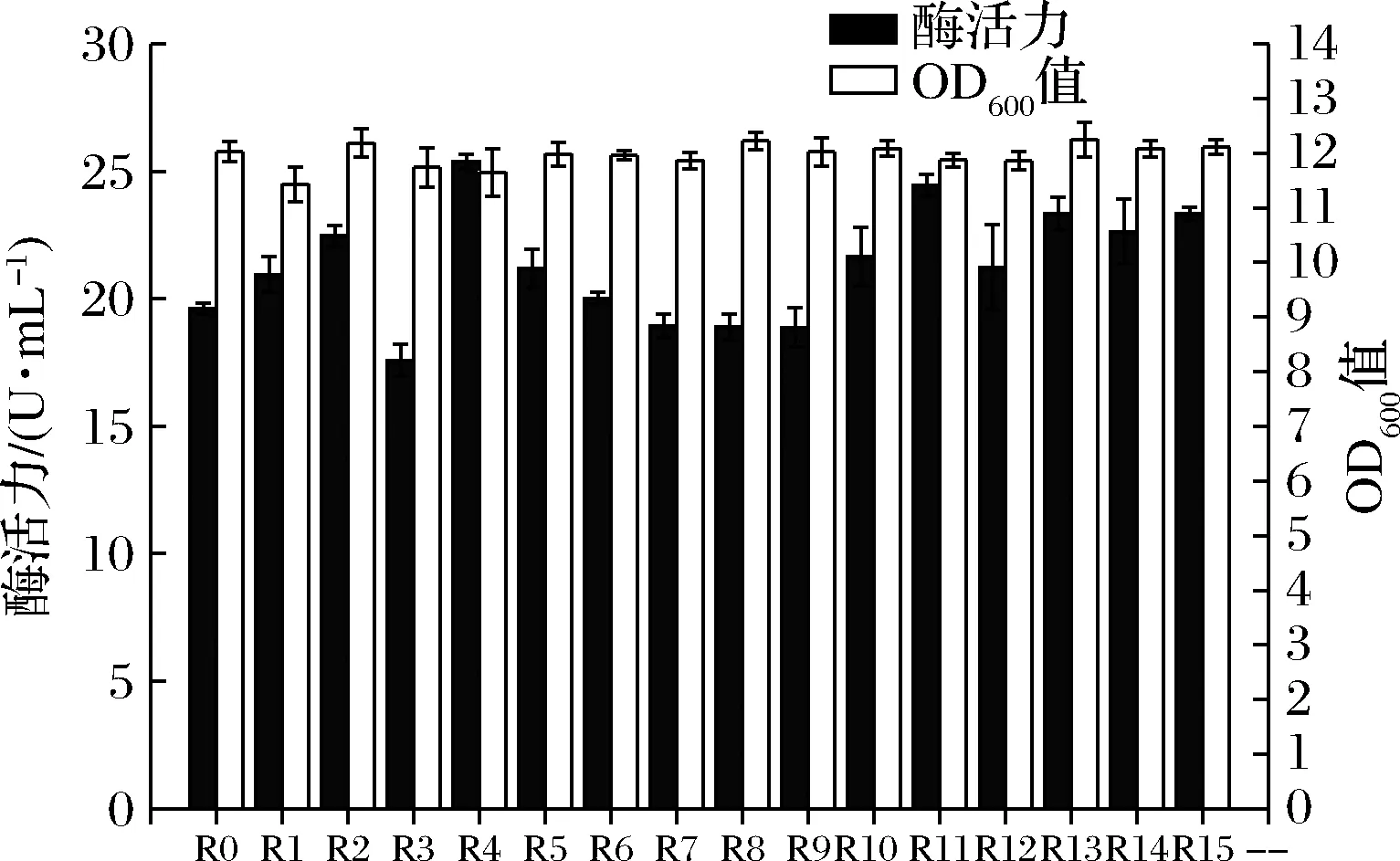
R0-原始对照菌株,R1~R15-RBS突变菌株(突变序列如图6所示)
3 结论
本文分别构建了9株单启动子和9株双启动子介导的DPE表达的重组菌株,其中单启动子Phag介导的重组菌株经摇床发酵后,最大酶活力高达19.62 U/mL,是P43介导的原始菌株酶活力的1.3倍,是枯草芽孢杆菌常用启动子PHpaII介导的重组菌株的1.77倍。然后通过RBS序列优化,构建了15株重组菌株,使其酶活力再次提高了29.4%,是原始菌株酶活的1.69倍,是PHpaII介导的重组菌株的2.29倍。综上所述,本研究为DPE工业化生产提供了方法学参考,后续可对其进行发酵优化,进一步提高其表达水平。
