掺锌生物活性玻璃纳米颗粒对复合树脂力学性能影响的实验研究
2022-10-02王路明曹潇仵琳悦李蕴聪雷波牛林
王路明 曹潇 仵琳悦 李蕴聪 雷波 牛林
1.西安交通大学陕西省颅颌面精准医学研究重点实验室 西安 710004;2.西安医学院第二附属医院口腔科 西安 710038;3.西安交通大学前沿科学技术研究院 西安 710054
复合树脂以其优异的美学效果及良好的力学性能在临床上广泛应用于牙体缺损修复[1],但与其他牙科修复材料相比,复合树脂容易产生聚合收缩,这种收缩会破坏充填体的边缘封闭性,在充填体与牙体组织之间形成微渗漏。此外,口腔微环境中的细菌容易在其表面黏附、聚集,形成菌斑生物膜,从而导致继发龋的产生[2]。为了克服这些缺陷,在复合树脂材料中添加具有抗菌生物效应的组分,研发能够抑制细菌黏附和生长的牙科复合树脂材料具有重要的临床意义。另外,预防釉牙本质脱矿的同时促进脱矿组织再矿化,也是减少继发龋发生,提高树脂充填修复成功率的有效策略[3-4]。近年来,生物活性玻璃以其良好的生物活性、生物相容性、骨传导性、组织矿化能力等在器官损伤修复[5]、癌症治疗[6]等方面的应用得到了广泛关注。锌元素是人体必需的微量元素之一,锌及其氧化物有着长效且安全稳定的抗菌特性[7]。若将两者的优势结合起来对复合树脂进行改性,增强复合树脂的抗菌性能和促进牙体组织再矿化的能力,将大大改善目前复合树脂性能的不足。本实验采用溶胶-凝胶模板法,在十二胺催化下制备掺锌生物活性玻璃纳米颗粒(Zn-doped bioactive glass nanoparticles,Zn@BGN),再用其对复合树脂改性,对改性后的复合树脂力学性能进行初步研究,同时筛选适宜的Zn@BGN添加比例,为进一步探讨Zn@BGN改性复合树脂的抗菌性能及再矿化能力提供依据。
1 材料和方法
1.1 实验材料仪器
1.1.1 主要原料去离子水、无水乙醇(C2H6O)、十二胺[dodecylamine;CH3(CH2)11NH2]、正硅酸乙酯(tetraethylorthosilicate,TEOS;C8H20O4Si)、磷酸 三 乙 酯(triethylphosphate,TEP;C6H15O4P)、四水硝酸钙[calcium nitrate tetrahydrate,CN;Ca(NO3)2·4H2O]、六 水 硝 酸 锌[zinc nitrate hexahydrate,ZN;Zn(NO3)2·6H2O],γ-甲基丙烯酰氧基丙基三甲氧基硅烷kh-570[γ-ethacryloxy propyl trimethoxyl silane;CH3CCH2COO(CH2)3Si(OCH3)3],以上材料均为萨恩化学技术(上海)有限公司产品。树脂体系采用双酚A双甲基丙烯酸缩水甘油酯(bisphenol A glycerolate dimethacrylate,BisGMA)和双甲基丙烯酸二缩三乙二醇酯(triethylene glycol dimethacrylate,TEGDMA)的混合物(Bis-GMA∶TEGDMA质量比为50∶50),0.7 μm硅化钡硼酸盐玻璃填料[silinated 0.7 micron Bbas(boron barium sulfate)glass],以上材料均为美国Esstech公司产品。引发体系包括光引发剂樟脑醌(camphorquinon,CQ)及光促进剂胺活化剂甲基丙烯酸二甲氨基乙酯[2-(Dimethylamino)ethyl methacrylate,DMAEMA],均为美国Sigma-Aldrich公司产品。
1.1.2 主要仪器分析天平(BSA2245型,Sartorius公司,德国),磁力恒温搅拌器(RH digital&ETS-D5型,IKA公司,德国),高速离心机(Multifuge X1型,Thermo公司,美国),涡旋振荡器(VoRTEX GENIUS 3型,IKA公司,德国),超声振荡器(KQ5200DE型,昆山超声仪器有限公司),冷冻干燥机(LABCONCO公司,美国),马弗炉(KSL-1200X型,合肥科晶材料技术有限公司),恒温振荡器(IS-RSD3型,CRYSTAL公司,美国),电热鼓风干燥箱(DHG-9140A型,上海一恒科学仪器有限公司),数显游标卡尺(500-196-30型,Mitutoyo公司,日本),光固化机(LED-F PLUS型,中国啄木鸟医疗器械有限公司),万能材料测试机(AGS-10kNG/500N型,岛津公司,日本),多功能数显显微硬度仪(MHVD-1000IS型,上海钜晶精密仪器制造有限公司),透射电子显微镜(transmission electron microscope,TEM;H7700型,HITACHI公司,日本),场发射扫描电子显微镜X射线光电子能谱仪(field emission scanning electron microscope/X-ray energy dispersive spectrometers,EDS;Quanta 250 FEG型,FEI公司,美国),X射线衍射仪(X-ray powder diffractometer,XRD;Bruker D8 ADVA-NCE型,Bruker AXS公司,德国)。
1.2 Zn@BGN的制备
使用溶胶-凝胶模板法,在十二胺催化下制备不同质量分数的Zn@BGN(表1)。将2 g十二胺、12.5 mL去离子水和40 mL无水乙醇加入烧瓶中,置于40℃的恒温磁力搅拌器上,充分搅拌15 min形成催化反应模板,先滴加500 μL TEOS,间隔10 min后再滴加500 μL TEOS,搅拌30 min后缓慢滴加103 μL TEP,继续搅拌30 min,按照表1所列的不同的掺锌质量分数(1.6%、2.6%、6.4%)依次加入已充分水解的CN和ZN,搅拌3 h后离心(10 000 r·min-1,15 min),去上清后加入无水乙醇洗涤3次,每次均使用超声振荡器及涡旋振荡器充分振荡后离心(10 000 r·min-1,5 min),去上清,再使用去离子水洗涤3次,得到生物活性纳米颗粒(bioactive glass nanoparticles,BGN)以及不同掺锌质量分数(1.6%、2.6%以及6.4%)的Zn@BGN凝胶,预冻后置入冷冻干燥机干燥,最后经过马弗炉600℃煅烧以去除结晶水和硝酸根,得到白 色BGN以 及3组 实 验 组Zn@BGN粉 末(1-Zn@BGN、2-Zn@BGN和6-Zn@BGN)。
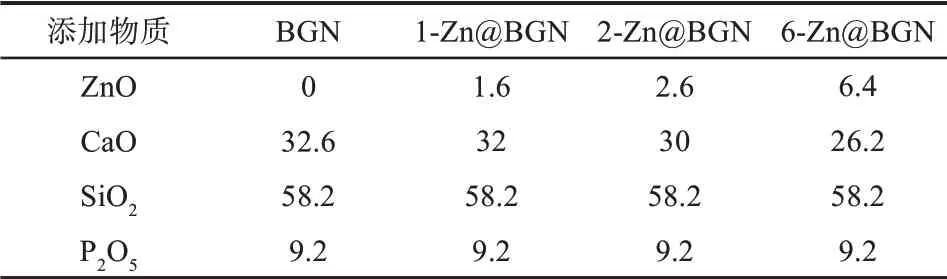
表1 不同掺锌比例BGN各组分质量分数Tab 1 Different proportion of Zn@BGN mass fraction of components %
1.3 Zn@BGN微观表征
取适量的BGN及3个实验组的Zn@BGN粉末,加入含有无水乙醇的小烧杯中,置于超声振荡器上振荡15~30 min,使之分散均匀。用毛细玻璃管吸取已振荡均匀的混合液,滴于铜网表面的碳支持膜上,确保样本均匀分布在支持膜上。待无水乙醇挥发完后,使用TEM对材料的形貌、粒径及分散性进行观察。另取适量样本,采用EDS进行面扫描,对样本元素成分进行测定。
1.4 材料体外生物活性检测
将BGN及Zn@BGN粉末按照1 g·L-1的质量浓度分别加入含有20 mL模拟体液(simulated body fluid,SBF)[8]的聚乙烯瓶中,将其固定在恒温振荡器中(120 r·min-1,37℃)持续摇动,经过10 d的反应后,过滤溶液,丙酮冲洗终止反应,去离子水洗涤,离心(10 000 r·min-1,5 min)3次,之后将其放入干燥箱中60℃干燥2 h获得反应后的材料。使用XRD对反应后材料进行连续扫描(10°~80°),XRD工作条件:Cu靶Kα射线,管电压40 kV,管电流100 mA。通过XRD图谱中呈现的磷灰石(hydroxyapatite,HA)特征峰判断材料的体外活性[9],筛选Zn@BGN中适宜的掺锌比例。将筛选出的掺锌比例适宜的Zn@BGN进行硅烷化处理,备用。
1.5 Zn@BGN改性复合树脂的制备
参照表2中复合树脂各组组分的配比,称取树脂体系(BisGMA/TEGDMA),避光环境下依次加入光引发体系(CQ和DMAEMA)、硅烷化的无机填料及1-Zn@BGN(通过上一步实验筛选得出)。制备对照组复合树脂和3个实验组复合树脂。对照组:未添加Zn@BGN改性树脂;实验组1:Zn@BGN-10改性树脂;实验组2:Zn@BGN-15改性树脂;实验组3:Zn@BGN-20改性树脂。
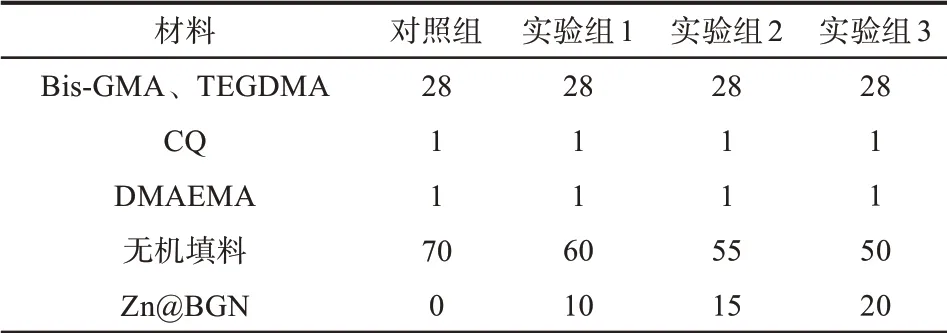
表2 实验用复合树脂各组分的质量分数Tab 2 The composite resin mass fraction of each composition %
1.6 复合树脂机械性能测定
1.6.1 挠曲强度(flexural strength,FS)依据ISO4049标准,用于FS测试试件的尺寸为长(l)25 mm,宽(w)2 mm,高(h)2 mm。将复合树脂充分填塞于定制的聚四氟乙烯磨具中,光固化灯灯头置于垂直于试件表面5 mm处,从前段、中段、后段依次照射,每个部位照射10 s,正反面相同(正面共30 s,反面共30 s),固化后脱模,打磨光滑,使用数显游标卡尺复测试件尺寸,37℃水浴中存储24 h,备用。使用万能材料试验机进行测试(跨距L为20 mm,速度为0.5 mm·min-1,传感器量程为10 kN),记录破坏载荷F。根据公式FS=3FL/2wh2计算FS。
1.6.2 径向拉伸强度(diametral tensile strength,DTS)依据美国国家标准,用于DTS测试试件的尺寸为直径(d)6 mm,高(h)3 mm。将复合树脂充分填塞于定制的聚四氟乙烯磨具中,光固化灯充分光照(正面30 s,反面30 s),固化后脱模,打磨光滑后,用数显游标卡尺复测试件尺寸,在37℃水浴中存储24 h,备用。使用万能材料试验机进行测试(速度为0.5 mm·min-1,传感器量程为10 kN),记录破坏载荷F。根据公式DTS=2F/πdh计算DTS。
1.6.3 压缩强度(compression strength,CS)依据美国国家标准,用于CS测试试件的尺寸为直径(d)4 mm,高(h)6 mm。将复合树脂充分填塞于定制的聚四氟乙烯磨具中,光固化灯充分光照(正面30 s,反面30 s),固化后脱模,打磨光滑后,用数显游标卡尺复测试件尺寸,在37℃水浴中存储24 h,备用。使用万能材料试验机进行测试(速度为0.5 mm·min-1,传感器量程为10 kN),记录破坏载荷F。根据公式CS=F/πr2(r=1/2d)计算CS。
1.6.4 维氏硬度(Vickers hardness,HV)依据ISO4049标准,用于HV测试试件的尺寸为长(l)6 mm,宽(w)5 mm,高(h)3 mm。将复合树脂充分填塞于定制的聚四氟乙烯磨具中,光固化灯充分光照(正面30 s,反面30 s),固化后脱模,测试面高度抛光至镜面状,其余面打磨光滑,用数显游标卡尺复测试件尺寸,37℃水浴中保存24 h,备用。使用多功能数显显微硬度仪对每个试件测试面取随机8个点进行测试,每个点加载力为0.98 N,保持5 s,得出各点的HV,计算每个试件的HV平均值。
1.7 统计学分析
本实验采用SPSS 22.0软件对数据进行统计学分析,经过方差齐性及正态性检验后,进行单因素方差分析,检验水准为双侧α=0.05。
2 结果
2.1 材料TEM表征
TEM微观检测显示:各组Zn@BGN粒径均在150 nm左右,形态规则,单分散性好(图1),与对照组BGN无明显差异。EDS元素分析显示结果与生物活性玻璃(SiO2-CaO-P2O5)相符(图2)。

图1 掺锌生物活性玻璃纳米颗粒TEM照片Fig 1 TEM photos of Zn@BGN
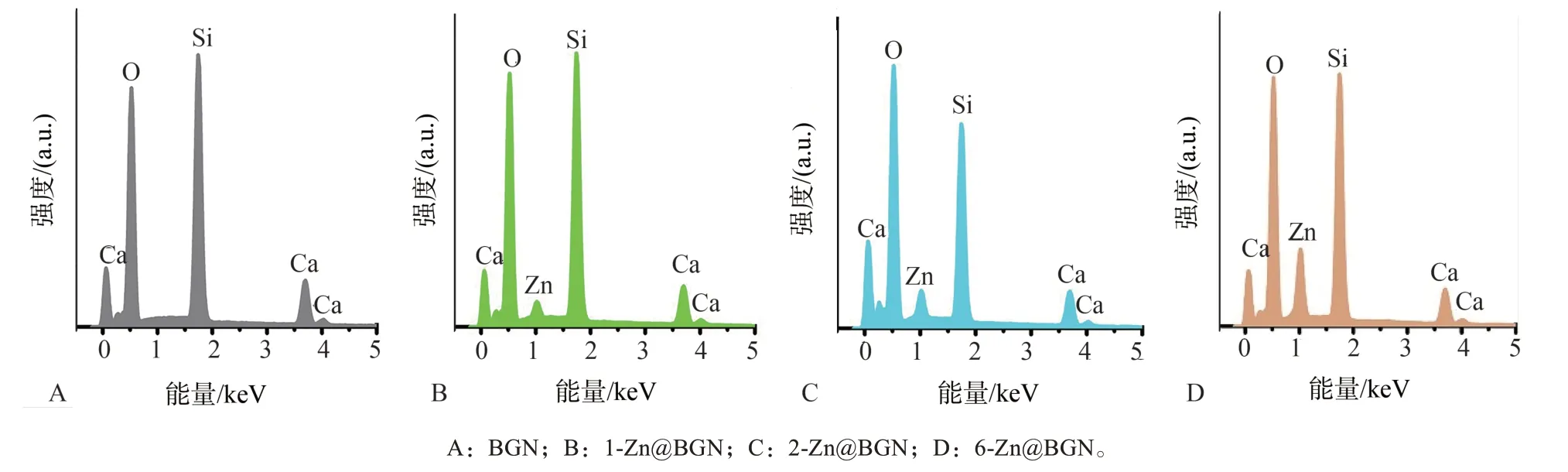
图2 掺锌生物活性玻璃纳米颗粒EDS元素分析图Fig 2 EDS element analysis diagram of Zn@BGN
2.2 体外生物活性
从XRD图谱中可以看出,BGN出现了HA特征峰,与2-Zn@BGN、6-Zn@BGN相比,当掺锌质量分数为1.6%(1-Zn@BGN)时,XRD图谱中也出现了HA特征峰(图3),由此通过体外HA实验筛选出了与BGN具有相同活性的合适掺锌比例。后续力学性能研究均采用1-Zn@BGN(以下简称Zn@BGN)进行测试。
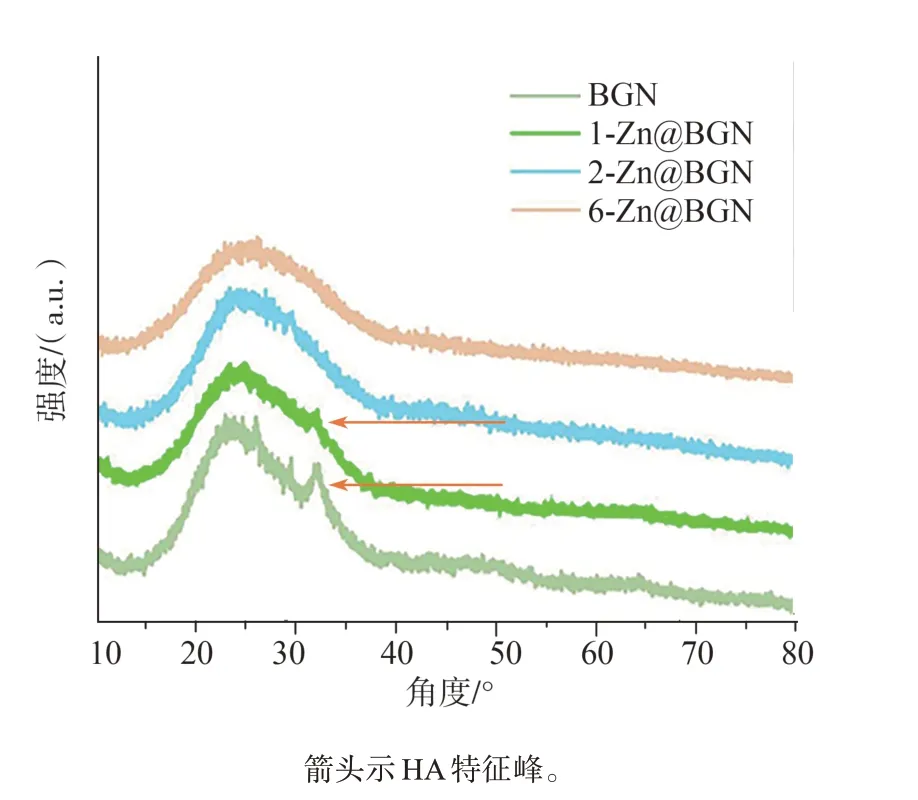
图3 XRD图谱中HA特征峰图Fig 3 XRD spectrum of HA characteristic peaks
2.3 Zn@BGN改性复合树脂力学性能
改性复合树脂力学性能测试结果见图4:Zn@BGN添加质量分数为10%和15%时,4项力学性能与对照组的差异均无统计学意义(P>0.05);当Zn@BGN的质量分数增加到20%时,FS和DTS性能下降,差异与对照组相比有统计学意义(P<0.05)。由此可以看出,Zn@BGN添加质量分数为15%及以下时,不影响复合树脂的力学性能。
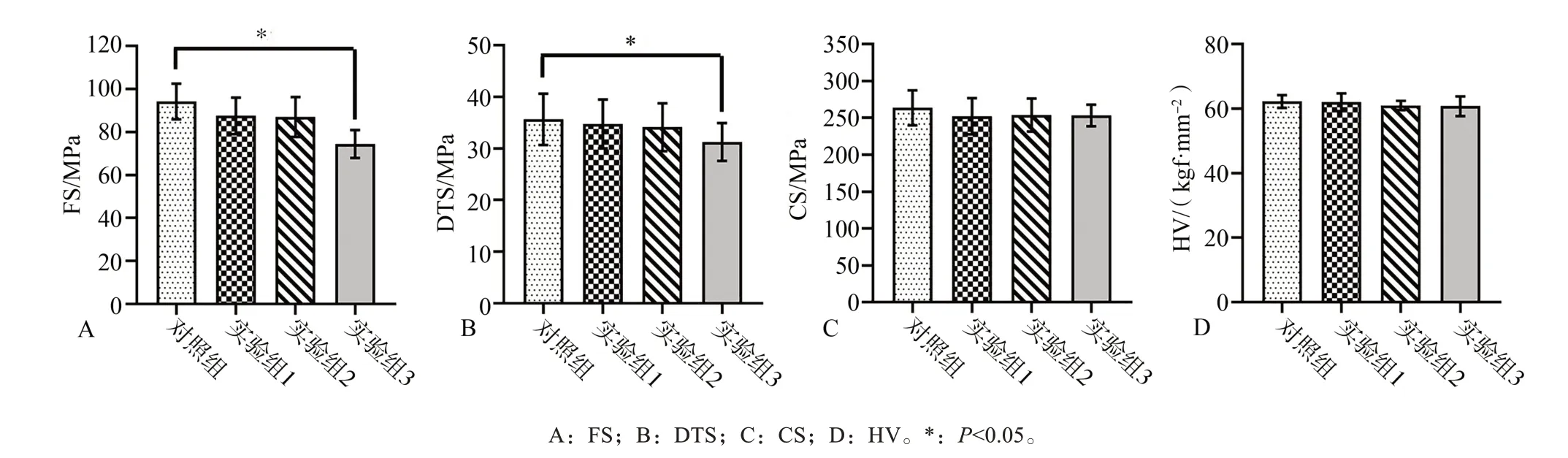
图4 改性复合树脂力学性能Fig 4 Mechanical properties of modified composite resin
3 讨论
20世纪60年代,Hench等研发了生物活性玻璃45S5,化学组分为Na2O-CaO-SiO2-P2O5[10]。因其具有良好的生物相容性和较高的骨修复能力[11],以及促进生物矿化及提高细胞活性等优越性能,在口腔医学领域已被广泛应用[12]。20世纪70年代Hench等又研制出熔融法来制备生物活性玻璃[13]。该方法类似于传统玻璃制备,即将一定量组成的原料混合,通过高温下熔融(1 300~1 500℃),最后淬冷得到最终产物。虽然该方法制备的生物活性玻璃在当时具有比较好的生物相容性及较高的骨修复能力,但其制备缺点十分明显。熔融法要求条件高,能耗大[14],得到的产物结构密实无孔[15],组成不均匀,比表面积较小[16]。直到20世纪90年代,Hench等又研制出了溶胶-凝胶法,并且成功制备58S、77S等生物活性玻璃,化学组分为SiO2-CaO-P2O5[17]。研究[15,18]表明,该方法的优点是可以在室温下制备,并可达到分子级别,大大提高了产品的结构均匀性,不会像熔融法所制备的那样密实无孔,而是具有大量的微纳米介孔,同时具有较高的比表面积和化学活性,从而提高了生物活性[12,15]。但此方法制备的生物活性玻璃因其颗粒容易发生团聚,严重影响了材料的分散性,即便通过后期筛分研磨[19-20]仍然欠佳,对其颗粒形态及尺寸的调控非常困难且繁琐。
本实验采用溶胶-凝胶模板法,在十二胺催化下制备出生物活性玻璃纳米颗粒,改善了之前合成方法的不足,而且十二胺同时具备模板剂和催化剂的双重作用[20]。通过十二胺的质量浓度可以控制生物活性玻璃的粒径和单分散性[20-21]。在本实验配置的十二胺质量浓度的催化下,制备出的BGN及Zn@BGN均呈现出规则的纳米球状颗粒,粒径约150 nm,大小较均一,单分散性良好(图1),得到了理想的生物活性玻璃纳米颗粒。原因考虑可能是由于十二胺的质量浓度不同导致了溶液碱性的变化[21]。在溶液碱性较低时,反应物的水解速率比较慢,在相同反应时间内颗粒不能迅速均匀长大,故无法形成均匀的球状颗粒;但随着溶液碱性的升高,反应物的水解及聚合速度同时加快,相同时间内水解的产物有更多的时间与周围的水解产物发生均匀碰撞,聚合增大,从而形成较均匀的纳米玻璃微球[8,20-22]。
复合树脂主要由树脂基质和硅烷化无机填料构成,其中无机填料的含量、颗粒的大小及形态、分布是否均匀均可对其机械性能产生影响[23],所以复合树脂中的填料并不见得越多越好。填料过多时会发生团聚,无法均匀分散,树脂连续相也同样会遭到破坏,使其各项力学性能指标下降。因此,常见的无机填料质量分数为60%~70%[24]。本实验中无机填料的总添加比例设定为70%,Zn@BGN则是对无机填料进行一定比例的替代。本实验中的BGN及Zn@BGN均已经过硅烷化处理,目的是提高与树脂基质的相容性[25],提升树脂的力学性能。已有研究[26]表明:树脂基质中掺入已硅烷化的生物活性玻璃,依然能较快速地释放出钙、磷等离子,诱导HA的生成。
力学性能测试结果显示:Zn@BGN添加质量分数为10%、15%时,FS、DTS、CS和HV与对照组的差异均无统计学意义,但添加质量分数增加到20%时,Zn@BGN改性复合树脂的FS和DTS出现了明显降低,差异有统计学意义。CS和HV虽然也有下降,但与对照组未见明显差异。FS和DTS降低的原因可能是纳米填料添加量达到20%后,树脂基质中出现了纳米颗粒团聚的现象,导致分布不均匀,从而出现树脂强度不一及树脂基质不均匀聚合收缩的情况[27],这种情况可导致应力容易集中在这些不均匀的部位从而产生裂纹,并且容易在这些部位扩展开来[28],影响其力学性能。在20%添加量下,CS和HV与对照组无明显差异,可能是因为CS和HV与FS和DTS测试原理不同所致。FS和DTS主要反应材料的韧性,以点线受力的方式测试,容易在分布不均的位置产生线性应力集中,造成断裂;而CS测试是将压力以面分散给整个柱形材料,不容易出现点或线的应力集中,所以产生的破坏没有FS和DTS反应的那么直接。此外,树脂的压缩性能本身要高于径向拉伸性能[29],这与本实验的结果有一致性。纳米颗粒的掺入会在复合树脂的表层形成纳米层,起初表面硬度会有所提高,但随着纳米材料掺入量的增加,表层的显微硬度会逐渐下降[30]。之所以HV与对照组未见差异,可能是由于Zn@BGN掺入量未达到出现表层硬度明显下降的拐点,而且无机填料的尺寸、组成及分布都有可能是影响硬度的因素[31]。
本实验采用溶胶-凝胶模板法,在十二胺催化下制备出了粒径大小均匀、单分散性良好且生物活性优异的Zn@BGN。利用其对复合树脂进行改性,Zn@BGN添加量为15%时,不影响改性复合树脂的力学性能,为进一步研究Zn@BGN改性复合树脂的抗菌性能及其再矿化能力提供了理论支持。
利益冲突声明:作者声明本文无利益冲突。
