Hf掺杂LaMnO3的第一性原理研究
2022-09-27吕书宇李建伟金浩卫亚东王健
吕书宇,李建伟,金浩,卫亚东,王健
深圳大学物理与光电工程学院,广东深圳518061
LaMnO3是带隙为1.10 eV 的A 型反铁磁(antiferromagnet,AFM)基态的钙钛矿半导体[1-3],空间群为Pbnm,由锰(Mn)以及氧(O)组成的八面体之间以氧为交点规则排列,如图1 所示,沿着垂直c轴的Mn的各层间构成反铁磁耦合.通过对La位原子进行替换掺杂,可以调控改变晶体内部的原子位置、电荷分布、自旋排列等多种参数,在复杂的相互作用下,产生诸如庞磁电阻效应[4]、铁磁相变[5-6]以及绝缘体-金属相变[7]等有趣物理现象.目前,使用+2价的钙(Ca)、锶(Sr)、钡(Ba)等对LaMnO3进行p 型掺杂的实验以及理论计算研究已经比较深入,但对于用+4 价的锑(Sb)、锆(Zr)、镨(Pr)和碲(Te)等对La进行替换的n型掺杂来说,由于氧空位的影响难以排除,氧化物中反常霍尔效应又影响了电子载流子的测量,使人们一直质疑是否形成了真正的n型导电机制.近年来,WANG等[8-9]使用价态较为单一的Hf 掺入LaMnO3中,成功合成了稳定的La1-xHfxMnO3,并通过X 射线光电子能谱分析,证明了La1-xHfxMnO3锰氧化物中Hf 是以Hf4+形式存在.本研究从密度泛函理论的第一性原理出发,研究不同掺杂浓度下La1-xHfxMnO3的晶体结构和电子结构,以及其光吸收性质和同质自旋阀器件.
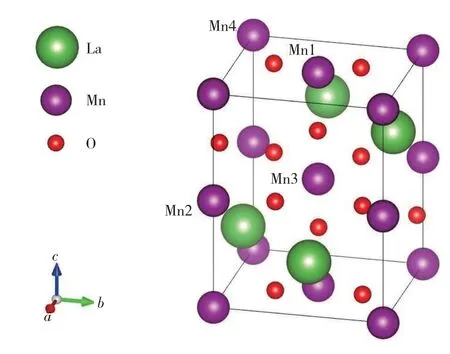
图1 正交晶系LaMnO3结构示意图(图中Mn1和Mn4与Mn2和Mn3分别位于相邻同一Mn层)Fig.1 The Schematic diagram of orthorhombic LaMnO3 structure.(Mn1 and Mn4 are in the same Mn layer perpendicular to the c-axis,while Mn2 and Mn3 are in the other nearest neighbor Mn layer.)
1 理论计算与LaMnO3基态确定
1.1 理论计算
本研究采用基于密度泛函理论的第一性原理计算软件包Vienna Ab-initio simulation package(VASP)进行结构计算,主要研究了锰氧化物钙钛矿LaMnO3以及其n 型掺杂La1-xHfxMnO3在不同掺杂浓度下的电子结构以及光吸收性质.在使用VASP 计算时,采用PBE 交换关联泛函[10],同时使用了投影缀加平面波(projector augmented-wave,PAW)赝势[11].计算中,Hf、Ba、La、Mn和O原子的价电子分别取为4 个电子(5d26s2)、10 个电子(5s25p66s2)、11 个电子(5s25p65d16s2)、7 个电子(3d54s2)和6 个电子(2s22p4).使用Hubbard-U方法修正锰氧化物钙钛矿中Mn的3d轨道电子间库伦作用,其中VASP软件中的ldautype参数设置为4.平面波截断能取为450 eV.使用共轭梯度法优化结构,获得弛豫晶格常数及所有原子位置.当掺杂浓度x≤0.125时,进行扩胞计算,当扩胞到2×2×2后,结构优化时不再弛豫扩胞的晶胞长度.采用8 ×8×6的网格做倒空间k点采样(扩胞后,对应扩展方向的k点数目将减少).晶格弛豫迭代过程中,设置原子间相互作用力的收敛标准为0.001 eV/nm,能量的收敛精度为1×10-5eV/原子.
为研究LaMnO3不同掺杂构成的同质结体系输运性质,采用基于NEGF-DFT的第一性原理计算软件包Nanodcal[12-13]计算了Ba 掺杂LaMnO3作为左右导线、Hf 掺杂LaMnO3作为中心层组成的自旋阀器件的磁电阻(magnetoresistance,MR),其中交换关联采用LDA +U方法,截断原子轨道基组采用Double-Zeta-Polarization,能量精度和截断能分别设置为1.0×10-5Hartree 和80 Hartree.计算透射系数时中心散射区k点设为6 × 6 × 1,导线偏压设为0 V.
1.2 LaMnO3基态
LaMnO3为过渡金属氧化物,在计算中需要引入自旋相反电子的强关联排斥能U,修正其Mn 原子中3d轨道电子的在位库伦能,使结果与实验相符 合.在VASP 中存在3种不同的加U方 法[14]:LIECHTENSTEIN 等[15]提出的旋转不变的局域自旋密度近似(local spin density approximation,LSDA)+U方法,DURAREV等提出的简化LSDA +U方法[16],以及类似第一种但不考虑LSDA 交换劈裂方法[17].为便于表述,本研究将这3种方法分别标记为LSDA +U-J、LSDA +(U-J)和LDA +U-J.其中,U为电子库伦相互作用项;J为交换相互作用项.LSDA+U-J方法与LSDA+(U-J)方法的区别在于前者的结果与U和J的取值都相关,而后者只与U-J相关.图2给出了3种加U方法下,反铁磁基态与铁磁基态的能量差ΔE=EAFM-EFM随着U值的变化.其中,J均设为0.5 eV.由图2 可见,3 种方法中,仅LDA+U-J方法给出LaMnO3的基态为反铁磁.在采用不同U的结构优化后,LDA+U-J方法下Ueff=U-J=5.0 eV 得到的晶格常数a=0.553 nm、b=0.586 nm 和c=0.768 nm 与实验结果[11-12]最一致.此外,3种加U下,Ueff=5.0 eV 得到的能带形状类似,带隙均约为1.10 eV[1].综合考虑,后续的计算中均采用LDA+UJ,并取Ueff=5.0 eV.

图2 LaMnO3反铁磁基态与铁磁基态的能量差随Ueff的变化(J ≡0.5 eV)Fig.2 The energy difference between the AFM and ferromagnetic(FM)state of LaMnO3 as a function of Ueff(J ≡0.5 eV).
2 Hf掺杂LaMnO3
2.1 掺杂LaMnO3磁基态
图3 给出了La1-xHfxMnO3和La1-xBaxMnO3在不同掺杂浓度x下的反铁磁基态能量与铁磁基态能量之差.在掺杂Ba 的情况下,随着掺杂浓度的上升,反铁磁与铁磁态能量之间的差值ΔEBa在x从0.1 升至0.2 时由负转变为正,意味着发生了反铁磁相向铁磁相的相变,这与实验观察的结果相符[18].从理论上说,当对LaMnO3进行p型掺杂(如掺Ca、Sr和Ba等元素)时,2 价阳离子替换了3 价阳离子,为使价态平衡,Mn4+的浓度随之升高.根据双交换作用机制[19],Mn4+与Mn3+之间的交换作用会倾向于铁磁耦合.在图3 中,随着Ba 掺杂浓度上升,ΔEBa也不断上升,意味着Mn3+与Mn4+的铁磁耦合逐渐占据主导地位.对于La1-xHfxMnO3来说,ΔEHf始终小于0,说明反铁磁态始终是La1-xHfxMnO3的基态.按照n型掺杂的情况,掺杂Hf 会使体系内价电子数增多,根据价态守恒,Mn2+的数量将不断增加,由于Mn2+的d轨道为半满模型,增强了阳离子与阴离子之间半价键交换作用[20],而半价键交换作用加深了Mn2+之间的反铁磁耦合程度,使得最终结果保持为反铁磁基态.
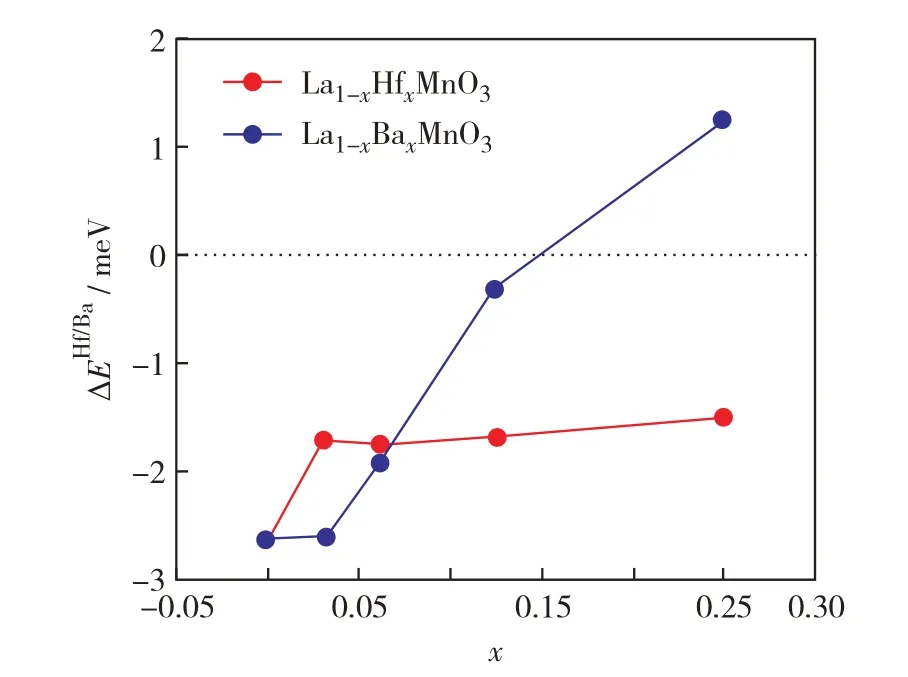
图3 La1-xHfxMnO3和La1-xBaxMnO3反铁磁态基态与铁磁态基态的能量差随着掺杂浓度的变化Fig.3 The energy difference between the AFM and FM state as a function of x in La1-xHfxMnO3(red line)and La1-xBaxMnO3(blue line).
2.2 晶体结构
当x为0.025、0.500和0.750时,以20个原子组成的Pbnm 空间群的LaMnO3原胞作为基本结构,并用Hf 分别替换1~3 个La 原子.当x为0.125、0.063、0.031、0.021 和0.014 时,分别将原胞进行 1 × 1 × 2、1 × 2 × 2、2 × 2 × 2、3 × 2 × 2 和3 × 3 × 2 等比例扩胞后再用Hf 替换1 个La 原子进行掺杂计算.结构优化时,当x≥0.031 时,扩胞的晶胞长度和原子位置一起弛豫;当x为0.021 或0.014时,由于掺杂浓度较低,只优化了原子位置,不变动扩胞的晶格常数.
表1列出了x从0增至0.750时各结构驰豫后扩胞长度折合到晶格常数的值,以及掺杂原子Hf 与最近邻O 原子间距和Mn—O—Mn 键角.由表1 可见,当x∈[0.031,0.750]时,扩胞长度折合后的晶格常数相比未掺杂结构发生了明显变化.比较晶格常数可以发现,随着x的上升,a和b长度逐渐变短,c逐渐变长,这意味着该正交晶系钙钛矿在a轴和b轴构成的ab平面内被压缩,而在c轴方向被伸长,原因是Hf 的离子半径小于La 的离子半径,在进行替换后,八面体之间的相互作用失去平衡,使钙钛矿结构产生畸变.

表1 La1-xHfxMnO3的晶格常数、Hf—O键长和Mn—O—Mn键角Table 1 The lattice constants,Hf—O bond lengths and Mn—O—Mn bond angles of La1-xHfxMnO3
2.3 电子结构
图4 为不同x下La1-xHfxMnO3的能带图.由图4可见,当x=0时,LaMnO3为半导体,带隙为1.10 eV,自旋向上和向下能带简并,与GONG等[1]的实验结果相符.在x=0.250、0.500、0.750 时,Hf的掺入对于能带结构的影响很大.当x=0.750时,75%的La 被Hf 替代,虽然体系仍是半导体,但其上下自旋能带不再简并,价带顶为自旋向上,导带底为自旋向下;当x=0.500时,体系变成了上下自旋简并的半导体,带隙为1.11 eV;而当x=0.250时,上下自旋能带简并再次解除,导带底和价带顶均由下自旋电子能级提供.这种现象源于Hf 的价电子改变了Mn原子在体系中的价态.在LaMnO3中Mn 离子为+3 价,3d 轨道上有4 个电子,按照洪德规则,这4个电子的自旋相同,锰氧化物钙钛矿的磁性也主要来源于Mn离子上3d轨道的电子自旋.
表2 列举了La1-xHfxMnO3中Mn 层的磁矩(m).由表2可知,当x=0时,各个Mn上磁矩的绝对值均为3.673μB(μB为玻尔磁子),构成LaMnO3的A型反铁磁结构,位于同一Mn层的Mn1和Mn4(图1)的磁矩为正,位于相邻Mn 层的Mn2 和Mn3 的磁矩为负,总磁矩为0.在替换1 个La 原子后,Mn1 和Mn4的磁矩几乎没有变化,而在负磁矩层中Mn2和Mn3的磁矩绝对值增加.其中,Mn2上磁矩的绝对值为3.837μB,比原来的3.673μB增加较小,而Mn3 上磁矩的绝对值变化较大,增加了约0.8μB,形成了亚铁磁半导体.因此,可以合理推测,替换1个La后,Hf4+会给体系多带来1个电子,这个电子通过交换作用基本转移到了Mn3 的3d 轨道上,令Mn3上磁矩的绝对值增大.而当替换2 个La 原子(x=0.500)时,多余的2 个电子分别转移到了不同Mn层自旋相反的Mn1 和Mn3 上(表2 第1 和第3 列),使得自旋能带再次简并,总磁矩为0.

表2 La1-xHfxMnO3中Mn的磁矩Table 2 Magnetic moment of Mn in La1-xHfxMnO3
当x为0.250~0.014 时,体系为20~360 个原子的LaMnO3超胞各用1 个Hf 替换1 个La 原子.当x<0.063 后,掺入Hf 比较少,在能带中间逐渐孤立出1 条掺杂导致的杂质能带,而带隙基本保持在~1.13 eV,见图4(f)至(h).

图4 La1-xHfxMnO3的能带图(a)x=0.750;(b)x=0.500;(c)x=0.250;(d)x=0.125;(e)x=0.063;(f)x=0.031;(g)x=0.021;(h)x=0.014;(i)x=0(红色为自旋向上的能带;蓝色为自旋向下的能带.)Fig.4 Band structures of La1-xHfxMnO3 for(a)x=0.750,(b)x=0.500,(c)x=0.250,(d)x=0.125,(e)x=0.063,(f)x=0.031,(g)x=0.021,(h)x=0.014,(i)x=0.(Red and blue lines represent the band structures with spin up and spin down states,respectively.)
为进一步探索La1-xHfxMnO3内部的物理机制以及掺杂带来的影响,以La0.969Hf0.031MnO3为例计算并分析其部分态密度(partial density of states,PDOS).图5 给出了电子能量E=[ -1.0,1.0] eV 时各元素的PDOS 组成.其中,PDOS >0的部分为上自旋态密度;PDOS <0的部分为下自旋态密度;费密能级设置在Ef=0 处.在图5 中-0.1 eV 处存在一个主要由Mn和O组成的只有下自旋的DOS,对应图4(f)的能隙中的孤立能级.进一步计算表明,该孤立态密度主要源自与Hf最近的8个Mn的其中之一,以及与其相近的O原子,该Mn的价态由+3变为+2价,磁矩绝对值也由3.680μB变为4.447μB.同时,无论掺杂浓度是多少,Hf 的磁矩始终≤0.01μB,因此,可认为Hf 的4 个价电子全部转移出去,Hf以+4价离子存在于体系中.

图5 La0.969Hf0.031MnO3中各元素的部分态密度图Fig.5 Partial density of states of Hf,La,Mn,and O in La0.969Hf0.031MnO3.
3 La1-xHfxMnO3光学性质
为探究La1-xHfxMnO3的光学性质,采用原子尺度材料模拟的计算机程序包(Vienna Ab-initio simulation package,VASP)计算介电函数,并使用式(1)计算光吸收系数.
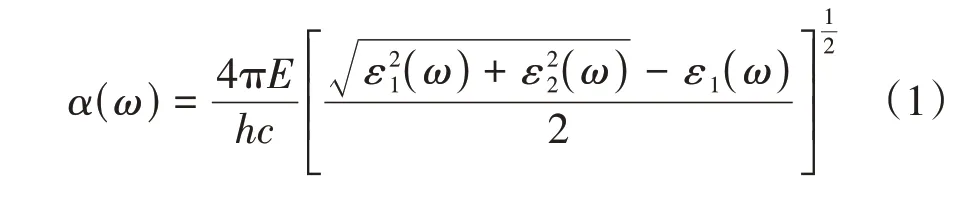
其中,ω为光照频率;ε1(ω)和ε2(ω)是介电函数的实部和虚部;h为普朗克常数;c为真空中的光速;E为电子能量.
图6 是利用式(1)计算得到的E在0~3.5 eV 时的La1-xHfxMnO3光吸收谱.对于本征LaMnO3即x=0时的结构而言,其光吸收带边大约位于1.3 eV 处,并且只有1 个位于2.2 eV 处的吸收峰.随着x从0增至0.125,光吸收边逐渐发生红移,而吸收峰的位置也缓慢左移,吸收峰的高度逐渐下降.当x=0.014、0.021和0.031时,光吸收边红移现象并不明显,但当x=0.063和0.125时,光吸收边发生较大偏离.
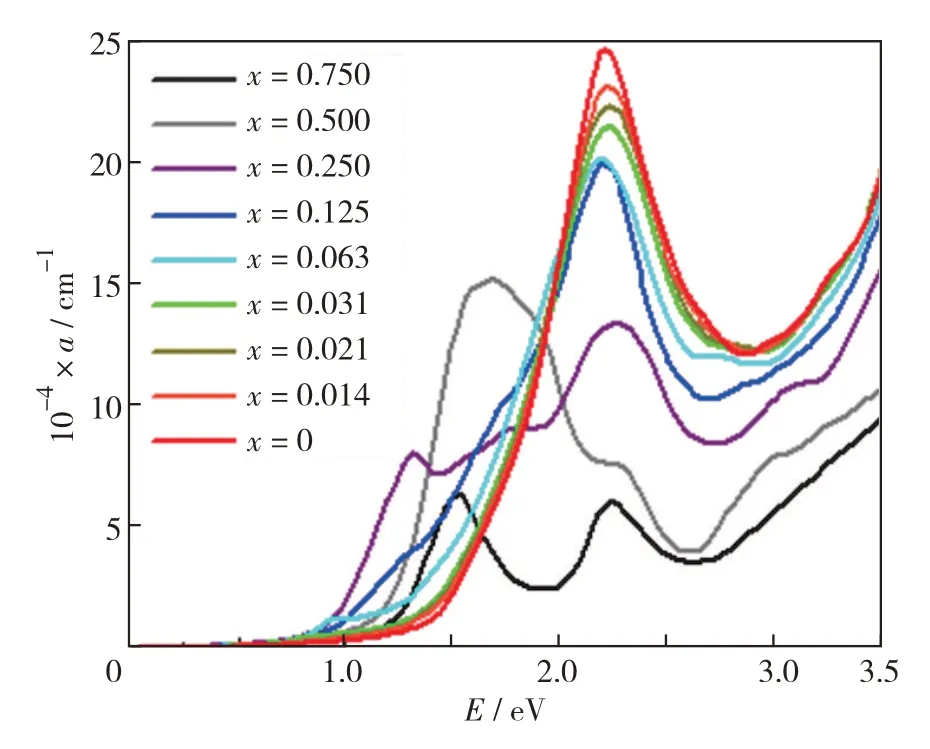
图6 La1-xHfxMnO3的光吸收系数图谱Fig.6 Absorption coefficient spectra of La1-xHfxMnO3.
当x=0.250、0.500 和0.750 时,由于结构变化较大,体系La1-xHfxMnO3的电子结构与LaMnO3相差较大,他们之间的光吸收也改变较大,吸收边发生大幅红移.当x=0.250和0.750时,光吸收曲线出现2个吸收峰;而x=0.500时,主要吸收峰又变为1 个.图4 能带图表明,x=0.250 和0.750 时,自旋劈裂,而x=0.500 时,自旋简并,说明光吸收图谱中的吸收峰与自旋劈裂的情况可能有关.
图7给出了不同掺杂浓度下La1-xHfxMnO3的光学带隙(Eoptg).由图7 可见,本征LaMnO3的光学带隙为1.28 eV[21],随着x的上升,La1-xHfxMnO3的光学带隙不断降低.其中,x分别为0.014、0.021、0.031、0.063、0.125 时对应的光学带隙分别为1.205、1.194、1.140、0.948和0.928 eV.可以发现,当x≥0.063 后,光学带隙下降明显.根据图4,当x在0.014~0.032 时,掺杂浓度较小,形成了孤立杂质能级,此时掺杂对体系的光学性质影响较小.而当x为0.063和0.125时,掺杂浓度变大,随之而来的是掺杂能级扩展成杂质能带,并使Hf逐步脱离掺杂地位,成为系统的主体元素之一,体系形成新的价带,使得带隙发生改变,从而显著改变了La1-xHfxMnO3的光学带隙.

图7 La1-xHfxMnO3的光学带隙Fig.7 Optical band gap of La1-xHfxMnO3.
3 LaMnO3掺杂体系在自旋阀中的应用
通过掺杂构造同质自旋阀具有很高的应用价值[22-23].在本研究中,同质LaMnO3自旋阀由Ba 掺杂LaMnO3作为左右导线,中间夹Hf掺杂LaMnO3的三明治构成.计算中,半无限长左导线和半无限长右导线由La3/4Ba1/4MnO3给出;中心散射区由左右两边各有1 层La3/4Ba1/4MnO3原胞作为缓冲区与导线相接,中间为2 层La3/4Hf1/4MnO3构成.图8 给出了中心散射区示意.当两边导线的Ba掺杂La3/4Ba1/4MnO3(包括缓冲层)与中间的Hf掺杂La3/4Hf1/4MnO3磁矩朝向相同时,自旋阀为平行结构(记为P),此时电导较高,而磁矩相反时为反平行结构(记为AP),电导较低,平行或反平行结构可通过对中间层和导线加同向或者反向磁场控制.对中心La3/4Hf1/4MnO3加门电压Vg进行调控,可以通过改变电子透射系数获得更好的自旋阀效果.

图8 由La3/4Hf1/4MnO3和La3/4Ba1/4MnO3组成的自旋阀中心散射区(青色球体为Ba;黄色为Hf;绿色为La;紫色为Mn;红色为O;箭头代表磁矩朝向;c方向为电子传输方向)Fig.8 The scattering region of the spin valve formed by La3/4Hf1/4MnO3 and La3/4Ba1/4MnO3,in which cyan,yellow,green,and purple represent Ba,Hf,La,and Mn elements,respectively.Arrow means magnetic direction and c is along the electron transportation direction.
费密能级电子透射系数T在P构型和AP构型下随门电压Vg的变化如图9(a).由图9(a)可见,随着Vg的升高,无论是P还是AP构型,T都会发生明显改变,当Vg处于-1.0~0.8 V 时,P 排列的电子透射率都高于AP 排列,且在Vg为0.3 eV 和0.6 eV 处分别存在2个峰值.
自旋阀的性能主要取决于费密能级处器件的磁电阻,计算式[24]为

其中,TP和TAP分别为P和AP构型的费密能级处的透射系数.由图9(b)可知,当Vg处于-1.0~0.2 V时,P 构型的透射系数比AP 排列大几个数量级,对应的MR 值非常高,有很好的自旋阀效果.在Vg=0 时,MR 值达到了~(2.8 × 105)%,在Vg=0.2 eV处可提升到~(5.1×105)%.

图9 (a)P和AP构型中费密能级处的透射系数;(b)费密能级处磁电阻MR随门电压Vg的变化趋势Fig.9 (a)Transmission coefficients for P and AP configurations,and(b)MR versus Vg at the Fermi energy.
结语
基于第一性原理通过计算研究了不同掺杂浓度La1-xHfxMnO3的晶格参数和电子结构.结果表明,La1-xHfxMnO3的基态为反铁磁基态或者亚铁磁基态,且能带结构皆为半导体,体现出n 型掺杂的性质.磁矩计算表明Hf 以稳定的+4 价出现,Hf4+替换La3+后,多余电子转移至相邻的1 个Mn 离子上.光吸收图谱表明,随着x的上升,La1-xHfxMnO3的光学带隙在不断下降,与能带结构密切相关.通过掺杂可以构成优异的同质LaMnO3自旋阀器件,器件磁电阻可达105%.
