舒悠颗粒质量控制研究*
2022-09-23屈小娟田元于吉臣刘建书刘玉凤赵玉珍冯会迎韩晓玲
屈小娟 田元 于吉臣 刘建书 刘玉凤 赵玉珍 冯会迎 韩晓玲**
(1.西安力邦制药有限公司,陕西 西安 710000;2.陕西功能食品工程中心有限公司,陕西 西安 710000;3.杨凌职业技术学院,陕西 杨凌 712100)
舒悠颗粒是由西安力邦制药有限公司与陕西功能食品工程中心有限公司合作开发的具有通便功能的保健食品。本产品选用质润多脂、润肠通便作用的火麻仁[1]和益气固、补气升阳作用的黄芪[2],辅以当归[3]、麦冬[4]补血活血、润肠通便,枳实[5]行气通腑、促进肠蠕动、弛缓肠平滑肌,沙棘[6]消食化滞、润肠通便,诸药并用,融润肠通便、补气固表、破气消积为一体,达到通便的目的。
便秘是指粪便在肠内滞留过久导致的秘结不通,排便周期延长,粪质干结,排出困难的情况[7]。中医学认为,便秘与胃、脾、肾等脏腑的功能失养,大肠传导失司相关[8],脾胃是全身气机升降的枢纽,调节支配消化系统,脾虚导致气行无力,无法驱动大小肠有效运动,导致便秘[9]。火麻仁能刺激肠粘膜分泌黏度,使肠道蠕动加快,减少大肠吸收水分[10],黄芪促进机体新陈代谢,增强小肠运动和平滑肌紧张度[11];当归明显改善血虚便秘症候,增加结肠和粪便含水量、软化粪便、改善结肠粘液的分泌,促进粪便排出[12-13];麦冬富含粘多糖,可促进大肠黏液的分泌,均具有润肠通便、缓解便秘的作用。另有研究[14]表明,沙棘中富含多糖和黄酮,而多糖和黄酮类物质具有明显的通便润肠作用。
本研究采用薄层色谱法(TLC)定性鉴别了舒悠颗粒中的当归、麦冬,采用紫外-可见分光光度法(UV-Vis)测定粗多糖和总黄酮的含量,采用高效液相色谱法(HPLC)测定黄芪甲苷的含量,以期控制舒悠颗粒的质量。
1 仪器与试药
1.1仪器 Agilent 1260高效液相色谱仪(安捷伦科技有限公司);TU-1810紫外-可见分光光度计(北京普析通用仪器有限责任公司);BSA224S电子天平(德国赛多利斯科学仪器有限公司);数显水浴锅(北京科伟永兴仪器有限公司);3-30K离心机(德国sigma);KQ5200DE数控超声波清洗器(昆山市超声仪器有限公司);WFH-203C紫外分析仪(上海精科实业有限公司)。
1.2试药 舒悠颗粒(批号:20171001、20171002、20171003,规格:3 g/袋);阴性样品1(不含当归)、阴性样品2(不含麦冬),均由陕西功能食品工程技术中心按舒悠颗粒的处方与制备工艺制得;当归对照药材(批号:120927-201617)、麦冬对照药材(批号:121013-201711)、黄芪甲苷对照品(批号:110781-201717,纯度:96.9%)、D-葡萄糖对照品(批号:110833-201707,纯度:99.9%)、芦丁对照品(批号:100080-201610,纯度:92.6%)均购自中国食品药品检定研究院;硅胶G薄层板、硅胶GF254薄层板均购自青岛海洋化工厂有限公司;乙腈为色谱纯,水为纯化水,其余试剂均为分析纯。
2 薄层色谱鉴别
2.1当归的薄层色谱鉴别 取本品粉末1.3 g,加乙醚50 mL,超声10 min,滤过,滤液蒸干,加乙醇1 mL溶解,作为供试品溶液。另取当归对照药材和当归药材各0.5 g,加乙醚20 mL,超声10 min,滤过,滤液蒸干,加乙醇1 mL溶解,分别作为当归对照药材溶液和当归药材溶液。按舒悠颗粒处方和工艺制备阴性样品1(不含当归),照供试品溶液制备方法制成阴性对照溶液。取上述四种溶液各10 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(4∶1∶0.1)为展开剂,预饱和30 min,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果,供试品色谱中,在与当归对照药材及当归药材在相应的位置显相同颜色的荧光斑点,专属性好,见图1。
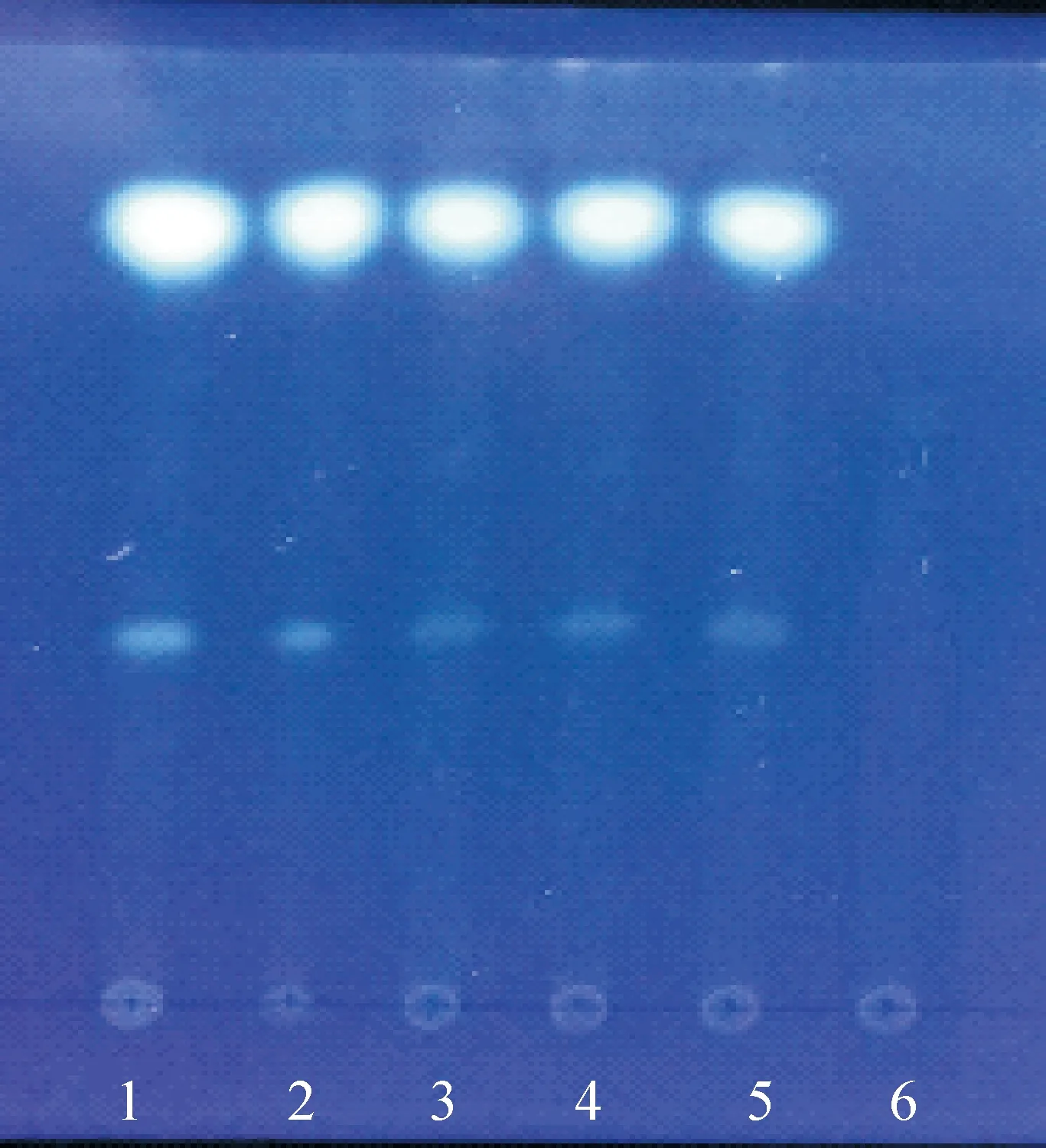
1.当归对照药材; 2.当归药材; 3.20171001号样品;
2.2麦冬的薄层色谱鉴别 取本品5 g,研成细粉,加水30 mL与盐酸3 mL,加热回流1 h,滤过,滤液用乙醚25 mL振摇提取,蒸干乙醚,加三氯甲烷2 mL溶解,作为供试品溶液。另取麦冬对照药材和麦冬药材各1 g,同法制成麦冬对照药材溶液和麦冬药材溶液。按舒悠颗粒处方和工艺制备阴性样品2(不含麦冬),照供试品溶液制备方法制成阴性对照溶液。分别取上述四种溶液各2 μL,分别点于同一硅胶GF254薄层板上,以正己烷-乙酸乙酯(1∶1)为展开剂,预饱和30 min,展开,取出,晾干,置紫外光灯(254 nm)下检视。结果,供试品色谱中,在与麦冬对照药材及麦冬药材在相应位置显相同颜色的荧光淬灭斑点,专属性好,见图2。

1.麦冬对照药材; 2.麦冬药材; 3.20171001号样品;
3 含量测定
3.1黄芪甲苷含量测定
3.1.1色谱条件 色谱柱:汉邦C18色谱柱(4.6×250 mm,5 μm);流动相:乙腈-水(32∶68);流速:1 mL·min-1;ELSD检测条件:漂移管温度:60 ℃;载气流量:1.6 L·min-1;增益:3.0。
3.1.2对照品溶液的制备 取黄芪甲苷对照品适量,精密称定,加80%甲醇溶解,稀释定容于5 mL量瓶中,混匀,配制成每1 mL含4.8489 mg黄芪甲苷的溶液,作为对照贮备液。精密吸取对照贮备液(4.8489 mg·mL-1)2.5 mL,置25 mL容量瓶中,用80%甲醇定容,摇匀,配制成每1 mL含484.89 μg黄芪甲苷的溶液,作为对照品溶液。
3.1.3供试品溶液的制备 取本品细粉约6 g,精密称定,置100 mL锥形瓶中,精密加入甲醇60 mL,超声(功率600 W、温度50 ℃)30 min,3000×g离心5 min。上清液滤过(0.45 μm微孔滤膜);滤渣用甲醇洗涤2次,每次30 mL,超声5 min,离心5 min,滤过,合并滤液,减压蒸干,加氨水溶液(10%)20 mL溶解,放置10 min,不时振摇,转移至分液漏斗中,用水饱和正丁醇萃取3次(30 mL、20 mL、20 mL)。合并萃取液,减压蒸干,残渣用80%甲醇溶解,转移至10 mL容量瓶中,加80%甲醇定容,摇匀,即得供试品溶液。
3.1.4阴性样品溶液的制备 按处方比例,取除去黄芪的其他药材,按照制剂工艺制成阴性样品,按照3.1.3项下供试品溶液制备方法制得阴性样品溶液。
3.1.5测定法 分别精密吸取标准溶液10 μL、20 μL,供试品溶液20 μL,注入液相色谱仪中,记录色谱图,用外标两点法对数方程计算。
3.1.6专属性 精密吸取对照品溶液、供试品溶液和缺黄芪阴性对照样品溶液各20 μL,分别注入液相色谱仪中,记录色谱图。对照品和供试品溶液里被测组分均能达到基线分离,且峰型对称,缺黄芪阴性对照样品中其他组分均不干扰黄芪甲苷测定,见图3。

A.黄芪甲苷对照品溶液;B.供试品溶液;C.缺黄芪阴性样品
3.1.7线性范围 精密吸取标准贮备液0.5 mL、1.5 mL、2.5 mL、3.75 mL、5 mL、7.5 mL,分别置25 mL量瓶中,用80%甲醇定容,摇匀。分别精密吸取20μL,注入液相色谱仪中,记录色谱图。以对照品浓度的对数为横坐标,以黄芪甲苷峰面积的对数为纵坐标,进行线性回归,黄芪甲苷在96.98~1454.66 μg·mL-1浓度范围内呈良好线性,回归方程为Y=1.761X-2.0411(r=0.9997)。
3.1.8定量限和检出限 以信噪比为3∶1对应的浓度为检出限,得黄芪甲苷检出限为40.03 μg·mL-1;以信噪比为10∶1对应的浓度为定量限,得黄芪甲苷定量限为120.09 μg·mL-1。
3.1.9精密度实验 精密吸取3.1.6项下线性测定中标4溶液20 μL,注入液相色谱仪中,重复进样6次,记录色谱图。黄芪甲苷峰面积RSD为2.38%(n=6),表明精密度良好。
3.1.10重复性实验 取本品,按3.1.3项下方法制备供试品溶液,共6份,分别精密吸取20 μL,注入液相色谱仪,记录色谱图,黄芪甲苷色谱峰面积RSD为1.16%,表明该方法重复性良好。
3.1.11稳定性实验 取本品,按3.1.3项下方法制备供试品溶液,分别在0 h、2 h、4 h、8 h、12 h、24 h时进样20 μL,记录色谱图。黄芪甲苷色谱峰面积RSD为0.46%,表明室温条件下供试品溶液在24 h内稳定性良好。
3.1.12回收率实验 精密称取本品(批号:20171001,黄芪甲苷含量为0.7136 mg·g-1)3 g,共9份,平均分为3组,每组分别加入浓度为4.8489 mg·mL-1的黄芪甲苷对照贮备液0.25 mL、0.5 mL、0.75 mL,按3.1.3项下方法制备供试品溶液,分别精密吸取20 μL,注入液相色谱仪,记录色谱图,计算回收率。平均回收率为95.55%,RSD为1.76%(n=6),回收率良好(见表1)。
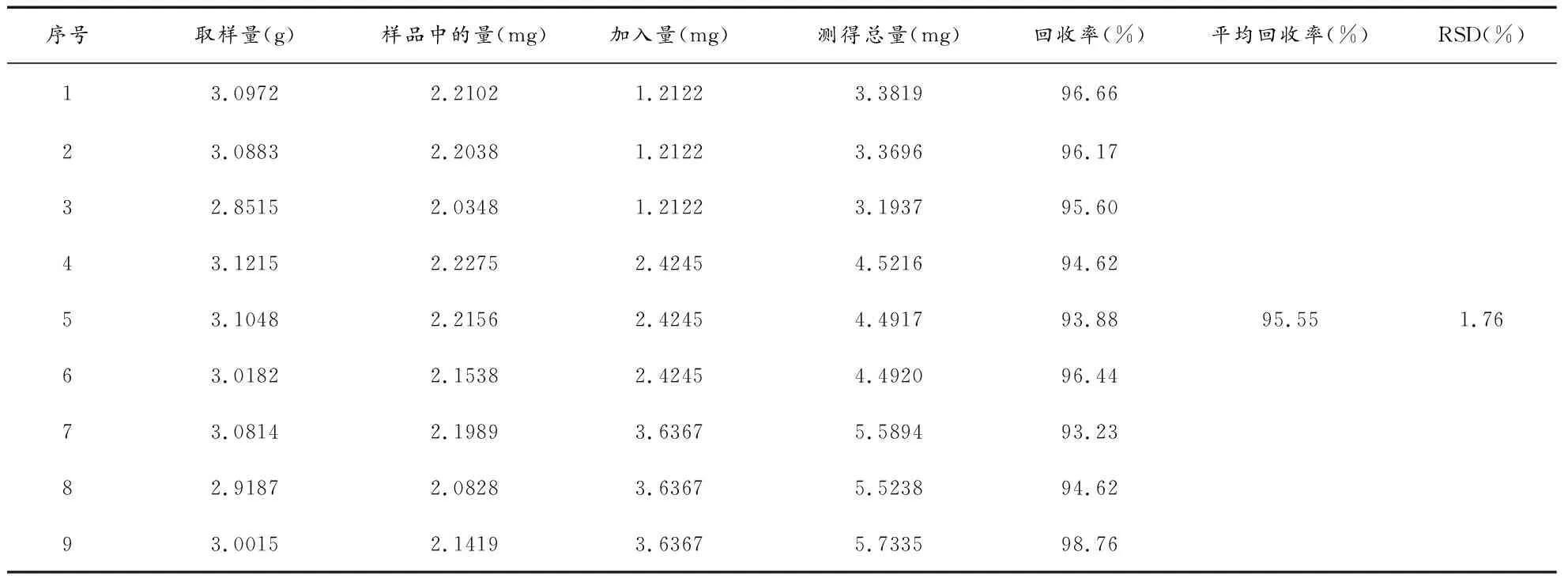
表1 回收率实验结果
3.1.13样品含量测定 取3批舒悠颗粒样品(批号为20171001,20171002和20171003)6 g,各两份,精密称定,照3.3.1项下操作,分别注入液相色谱仪,记录色谱图。3批舒悠颗粒中黄芪甲苷含量分别为0.7107 mg·g-1、0.7041 mg·g-1、0.7059 mg·g-1(见表2)。
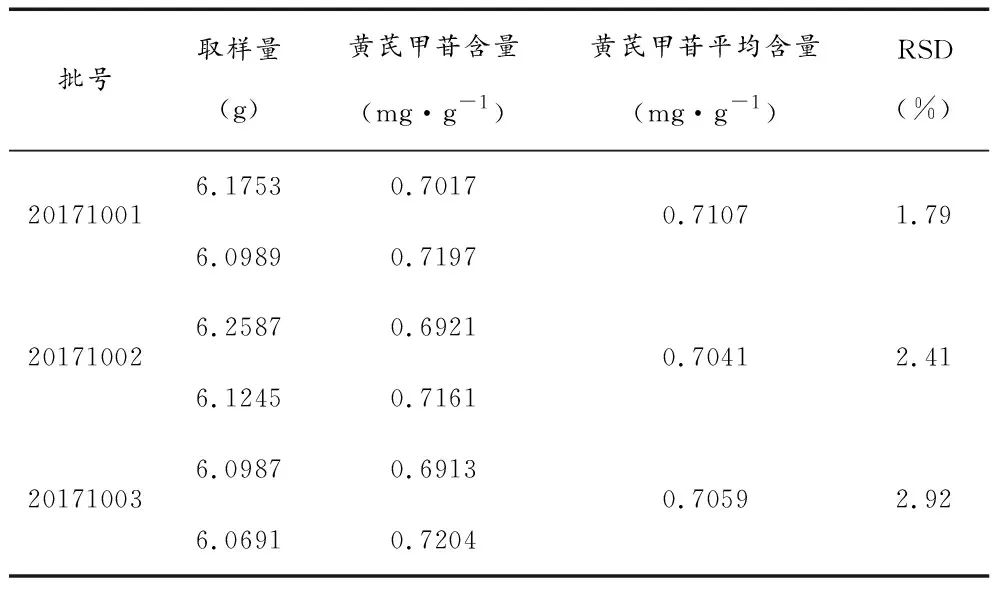
表2 黄芪甲苷含量测定结果
3.2粗多糖含量测定
3.2.1供试品溶液的制备 取本品0.5 g,精密称定,加乙醚100 mL,40 ℃加热回流1 h,静置,放冷,小心倾去乙醚,残渣于水浴上蒸干。加80%乙醇100 mL,80 ℃加热回流1 h,趁热滤过,滤渣与滤器用热80%乙醇30 mL分次洗涤,滤渣连同滤纸置烧瓶中,加水150 mL,100 ℃加热回流2 h。趁热滤过,用少量热水洗涤滤器,合并滤液和洗液。加糖化酶30 mg,于60 ℃、pH4.5条件下酶解1.5 h,小心加热至沸,冷却,过滤,滤液加水至150 mL,取30 mL,置250 mL锥形瓶,加无水乙醇200 mL,混匀,于4 ℃冰箱静置4 h以上,6000 rpm离心15 min,弃去上清液,残渣用水溶解、定容至50 mL,混匀。
3.2.2标准曲线的绘制 精密称取D-葡萄糖对照品10.01 mg,置100 mL容量瓶中,加水适量溶解,稀释至刻度,摇匀,制成每1 mL中含D-葡萄糖0.10 mg的溶液,作为葡萄糖对照贮备液。精密量取葡萄糖对照贮备液0 mL、0.2 mL、0.4 mL、0.6 mL、0.8 mL、1.0 mL、1.2 mL,置10 mL具塞试管中,加水至2.0 mL,精密加入5%苯酚溶液1 mL,摇匀,迅速精密加入硫酸5 mL,摇匀,放置10 min,置100 ℃水浴中2 min,取出,迅速冷却至室温,以相应试剂为空白,于485 nm处测定吸光度。以吸光度(A)为纵坐标,葡萄糖的浓度(μg·mL-1)为横坐标,进行线性回归,得线性方程。
3.2.3测定法 精密量取供试品溶液1 mL,置10 mL具塞试管中,加水1 mL,照3.2.2项下自“精密加入5%苯酚溶液1 mL”起,至“于485 nm处测定吸光度”操作。由线性方法计算供试品溶液的浓度C,计算样品中粗多糖含量。
3.2.4线性范围 照3.2.2项下操作。葡萄糖在0~15.00 μg·mL-1的浓度范围内呈良好线性,标准曲线方程为:A=0.0466×C+0.0068,相关系数r=0.9996。
3.2.5检出限 以20次空白测定值的3.3倍标准偏差除以标准曲线斜率(即LOD=3.3δ/S)计算,粗多糖的检出限为0.4 μg·g-1,表明该法检出限低,灵敏度高。
3.2.6精密度实验 精密吸取葡萄糖标准溶液(0.10 mg·mL-1)0.6 mL,加水1.4 mL,共6份,照3.2.2项下自“精密加入5%苯酚溶液1 mL”起,至“于485 nm波长处测定吸光度”操作。分别测定,葡萄糖吸光度RSD为0.10%(n=6),表明仪器精密度良好。
3.2.7重复性实验 取本品0.5 g,共6份,按3.2.1项下方法制备供试品溶液,分别测定。粗多糖平均含量为33.634 mg·g-1,RSD为1.96%,表明本方法重复性好。
3.2.8稳定性实验 取供试品溶液,分别于0 h、2 h、4 h、8 h、12 h、24 h时按3.2.3项下测定,记录吸光度。吸光度的RSD为1.64%,表明室温条件下供试品在24 h内稳定。
3.2.9回收率实验 取本品(批号:20171001,粗多糖含量为33.634 mg·g-1)0.25 g,精密称定,共9份,平均分为3组,每组分别加入浓度为1.00 mg·mL-1的葡萄糖标准贮备液4 mL、8 mL、12 mL,照3.2.1及3.2.3项下操作,记录吸光度,计算回收率。平均回收率为99.04%,RSD为1.35%(n=6),表明回收率良好(见表3)。
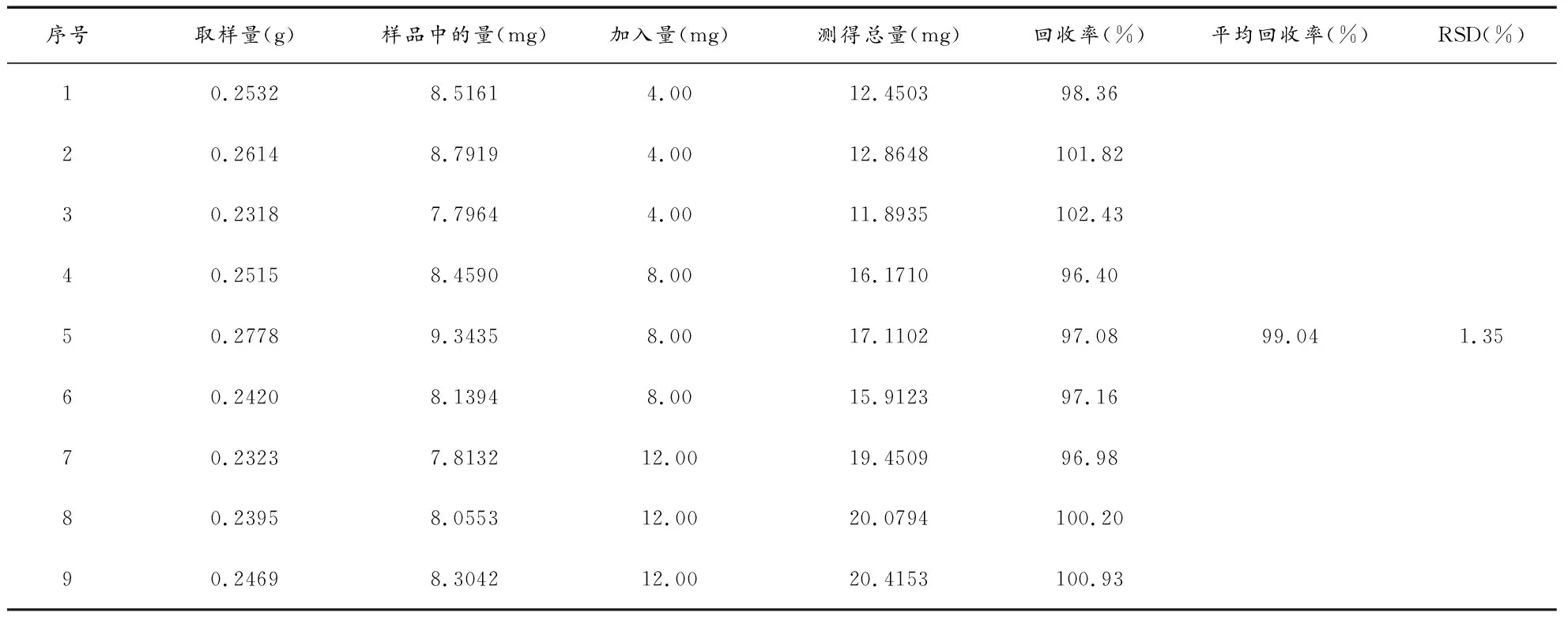
表3 回收率试验结果
3.2.10样品含量测定 取3批舒悠颗粒样品(批号为20171001,20171002和20171003)0.5 g,各两份,精密称定,照3.2.1及3.2.3项下操作,记录吸光度。3批舒悠颗粒样品中粗多糖含量分别为33.749 mg·g-1、35.105 mg·g-1、33.302 mg·g-1(见表4)。
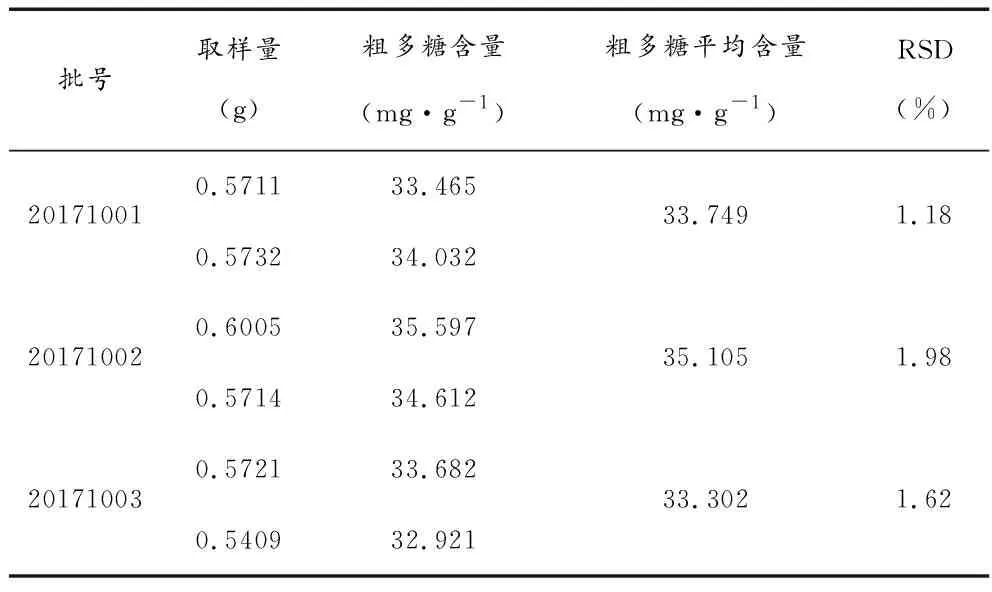
表4 粗多糖含量测定结果
3.3总黄酮含量测定
3.3.1供试品溶液的制备 取本品0.25 g,精密称定,加乙醇定容至25 mL,摇匀,超声提取20 min,静置,取上清液1.0 mL于蒸发皿中,加聚酰胺粉1 g,于水浴上挥去乙醇,转入层析柱。加石油醚(30~60 ℃)20 mL淋洗,弃去淋洗液,加甲醇20 mL洗脱,收集洗脱液并定容至25 mL,混匀。
3.3.2标准曲线的绘制 取芦丁对照品适量,精密称定,加甲醇溶解,定容至100 mL,摇匀,制成每1 mL含芦丁47.60 μg的溶液,作为芦丁对照贮备液。取芦丁对照贮备液0 mL、1.0 mL、2.0 mL、3.0 mL、4.0 mL、5.0 mL于10 mL比色管中,加甲醇至刻度,摇匀,于360 nm处测定吸光度。以吸光度(A)为纵坐标,芦丁的浓度(μg·mL-1)为横坐标,进行线性回归,得标准曲线方程。
3.3.3测定法 取供试品溶液,于360 nm处测定吸光度,代入标准曲线方程计算总黄酮含量。
3.3.4线性范围 照3.3.2项下操作。芦丁在0~23.80 μg·mL-1浓度范围内呈良好线性,标准曲线方程为:A=0.0261×C+0.0006,相关系数r=0.9994。
3.3.5检出限 以20次空白值的3.3倍标准偏差除以标准曲线斜率(即LOD=3.3δ/S)计算,总黄酮的检出限为1.8 μg·g-1,表明该法检出限低,灵敏度高。
3.3.6精密度实验 精密吸取芦丁对照贮备液(47.60 μg·mL-1)3.0 mL于10 mL比色管中,加甲醇至刻度,摇匀,于360 nm处测定吸光度。芦丁吸光度RSD为0.11%(n=6),表明该法精密度良好。
3.3.7重复性实验 精密称取本品0.25 g,共6份,照3.3.1及3.3.3项下操作,分别测定吸光度,总黄酮RSD为2.14%,表明该法重复性良好。
3.3.8稳定性实验 取供试品溶液,分别于0 h、2 h、4 h、8 h、12 h、24 h时照3.3.3项下操作,记录吸光度。吸光度RSD为2.01%,表明室温条件下供试品溶液在24 h内稳定。
3.3.9回收率实验 精密称取本品(批号:20171001,总黄酮含量为1.6476 mg·g-1)0.2 g,共9份,各置25 mL量瓶中,平均分为3组,每组分别加入浓度为0.20 mg·mL-1的芦丁标准贮备液0.5 mL、1.0 mL、1.5 mL,照3.3.1及3.3.3项下操作,记录吸光度,计算回收率。平均回收率为98.47%,RSD为1.83%(n=6),表明该方法回收率良好(见表5)。
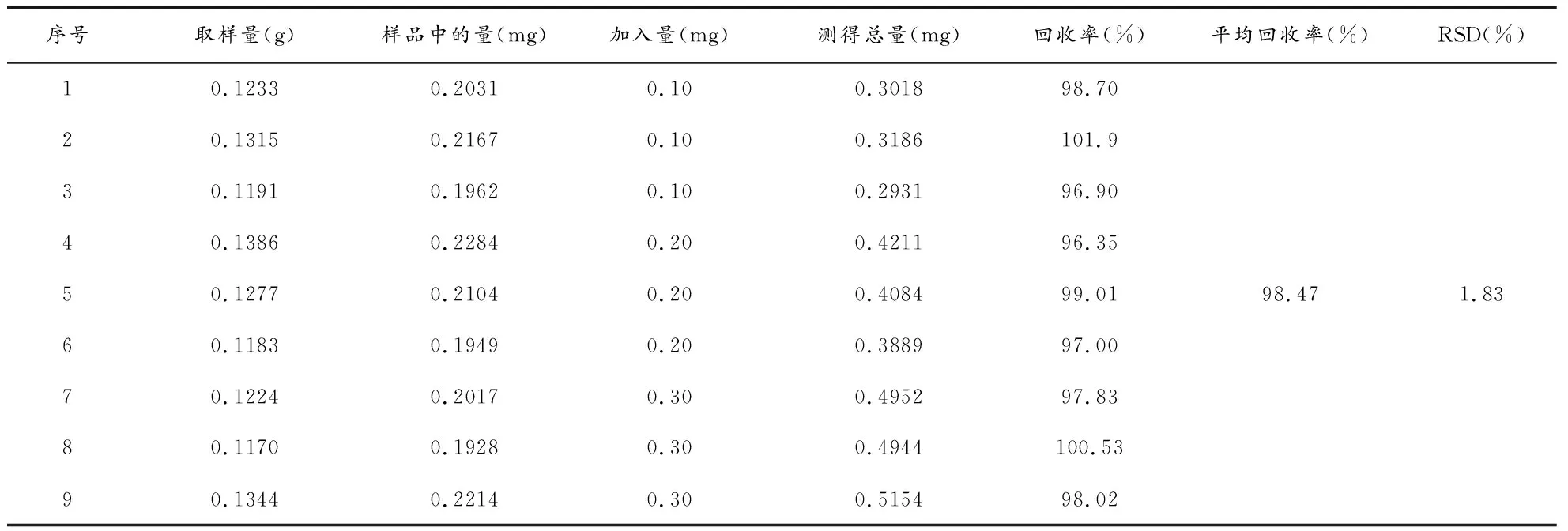
表5 回收率实验结果
3.3.10样品含量测定 精密称取3批舒悠颗粒样品(批号:20171001,20171002,20171003)0.25 g,各两份,精密称定,照3.3.1及3.3.3项下操作,记录吸光度。3批舒悠颗粒样品中总黄酮含量分别为1.6042 mg·g-1、1.5955 mg·g-1、1.5837 mg·g-1(见表6)。
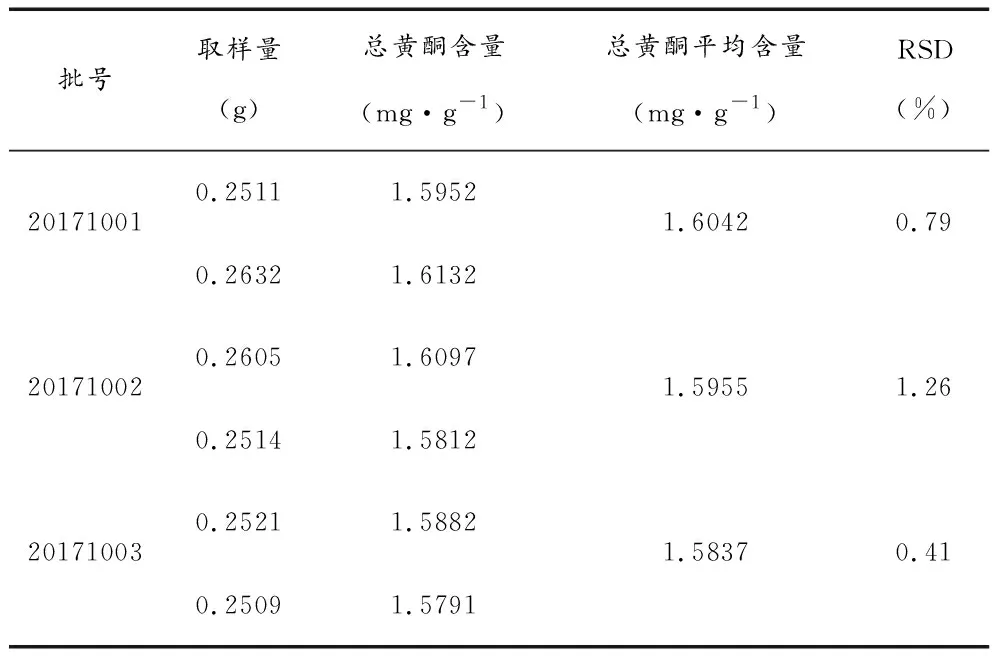
表6 总黄酮含量测定结果
4 讨论与小结
4.1薄层色谱分析
4.1.1当归的薄层色谱鉴别 本研究参照《中国药典》2015年版一部[15]当归项下薄层色谱方法,以正己烷-乙酸乙酯(4∶1)为展开剂试验,薄层色谱中舒悠颗粒样品未显示荧光斑点;参考相关文献和资料[16-17],对展开剂进行了优化研究,分别采用甲苯-乙酸乙酯-甲酸(4∶1∶0.1)、正己烷-乙酸乙酯(9∶1)、石油醚(60~90℃)-乙酸乙酯(19∶1)和甲苯-乙酸乙酯-甲酸(20∶10∶1)等展开剂实验。结果表明,展开剂为甲苯-乙酸乙酯-甲酸(4∶1∶0.1)时,在当归对照药材及当归药材相对应的位置上,舒悠颗粒样品的薄层色谱图显现清晰的荧光斑点,且与其它斑点分离良好。
4.1.2麦冬的薄层色谱鉴别 采用《中国药典》2015年版一部[15]麦冬项下的薄层色谱方法试验,得到的薄层色谱图未出现特征斑点。参考相关文献[15,18-19],分别对供试品溶液的制备方法、展开剂组成及比例进行了优化。结果表明,采用中药制剂妇宝颗粒的供试品溶液制备方法和展开剂正己烷-乙酸乙酯(1∶1)可得到清晰斑点及较好的分离效果。优化后的方法缩短了样品前处理时间,且不用甲苯、三氯甲烷等有毒试剂。
4.1.3其他成分的薄层色谱鉴别 笔者还对火麻仁、枳实、沙棘等进行了定性鉴别研究,采用薄层色谱鉴别方法,通过优化供试品溶液制备方法、展开剂组成和比例、展开温度、湿度,均未得到清晰的色谱斑点、分离效果不佳。因此,暂不将火麻仁、枳实和沙棘列入质量标准。
4.2含量测定分析 考虑到君药火麻仁无特定的指标成分,无法直接反映制剂质量,因此选用黄芪甲苷、粗多糖、总黄酮等有效成分含量进行研究。
4.2.1黄芪甲苷含量测定 黄芪甲苷常用的检测方法有HPLC-UV、HPLC-ELSD、薄层扫描法等。因黄芪甲苷仅在紫外末端有吸收,采用紫外检测器检测灵敏度低,干扰大,重现性差;《中国药典》2015年版的检测方法(HPLC-ELSD法),费时费工,样品制备繁琐,且未规定明确的蒸发光检测参数;薄层扫描法稳定性、重现性差,操作繁琐。为此,参考相关文献,通过一系列的实验优化,建立了一种简便快速、准确的黄芪甲苷含量测定方法。
笔者对比研究了超声和加热回流两种提取方法的提取效率,结果表明无明显差异;对超声工艺参数(超声功率、超声时间、温度)进行了优化,在超声时间30 min、温度50 ℃、超声功率600 W条件下黄芪甲苷的提取率最高,采用此提取方法,提取时间从《中国药典》2015年版方法中的4 h缩短到30 min。
参照《香港中药材标准(第一册)》[20]中黄芪项下黄芪甲苷含量测定方法中供试样品处理方法,对黄芪甲苷富集纯化方法进行了优化,采用氨试剂溶解提取物,再用水饱和正丁醇萃取,去除酚、酸性成分等,以达到富集纯化的目的。与《中国药典》2015年版测定方法相比,简化了操作步骤,省去了大孔树脂洗脱步骤,缩短了分析时间,降低了因操作冗繁带来的实验误差。
研究了蒸发光检测器的漂移管温度和雾化器气体流量对黄芪甲苷响应值的影响,最终选择漂移管温度60℃、载气氮气的流速1.6 L·min-1、增益3.0检测方法,获得了较好的分析效果。
通过以上研究,简化了黄芪甲苷含量测定的操作步骤,缩短了分析时间,提高了分析精确性。
4.2.2粗多糖含量测定中供试品溶液制备方法的优化 舒悠颗粒中添加了辅料糊精,需用糖化酶将糊精酶解为单糖以排除干扰。本研究对糖化酶添加量(25 mg、30 mg、35 mg)和酶解时间(1 h、1.5 h、2 h、2.5 h)进行了优化,用碘液测试是否酶解完全。结果表明当糖化酶添加量为30 mg,酶解时间1.5 h酶解完全,可确保检测结果的准确性。
综上,本研究建立的当归、麦冬的薄层色谱鉴别方法以及黄芪甲苷、粗多糖、总黄酮的含量测定方法,专属性强、方法简单可行、精密准确,适用于舒悠颗粒的质量控制。
