ZC4H2 基因c.82G>A 杂合半合子错义变异致ZC4H2相关罕见疾病的基因诊断(附1例分析)
2022-09-21李沙沙崔清洋周福军贾倩芳
李沙沙,崔清洋,周福军,贾倩芳
新乡医学院第一附属医院儿科,河南卫辉 453100
先天性多关节挛缩(AMC)是一种罕见的神经肌肉疾病,发病率为1/5 000~1/3 000,其特点是至少涉及两个关节的非进行性先天性挛缩。与AMC相关的潜在遗传原因和发病机制多种多样,尚未完全了解[1]。近年来,包括外显子组和基因组研究在内的新一代测序技术研究显示,与AMC 相关的基因超过400个,AMC与常染色体显性遗传、常染色体隐性遗传和 X 连锁遗传有关[2-3]。ZC4H2 基因致病变异可导致一种罕见、特殊的X-连锁AMC 性疾病,即Wieacker-Wolff 综合征(WWS),也称为 ZC4H2 相关罕见疾病(ZARD),为X连锁隐性遗传的神经发育障碍,该病于1985年首次报道[4],至今文献报道病例很少,与ZC4H2 基因变异相关的基因型—表型谱也非常有限[5]。ZARD 的临床特征包括 AMC、智力障碍和运动发育迟缓[6],男性患者的临床症状比女性患者更严重,部分女性患者也可出现比较严重的症状,尤其是ZC4H2 基因缺失和无义变异病例,部分病例被认为与非随机X 染色体失活(XCI)有关[2]。ZARD国内报道甚少,现对 1 例 ZC4H2 基因 c.82G>A 杂合半合子错义变异致ZARD 患儿的临床资料进行回顾性分析,以期加强临床儿科医师对本病的认识。
1 资料分析
患儿女,7 个月18 天,因发育落后3 个月入院。4月龄时家长发现患儿不会抬头,未予重视。7月龄时患儿抬头较前稍好转,但仍竖头不稳,收住入院。患儿系第1 胎第1 产,足月顺产,无窒息及抢救史。患儿父母均体健,非近亲结婚。体格检查:体质量9.4 kg,头围43 cm,身长66 cm,抱入病房,发育落后,双眼内斜视,颈短但无转动困难,心肺腹未见异常,双手掌指关节挛缩,双手拇指尺偏,双足底呈摇篮底足,双下肢肌张力增高。仰卧位:头可居中线,追视、追听可,双手偶可居中线活动,双下肢自然伸展;俯卧位:主动肘支撑,抬头90°,时间短;坐位及前倾坐位:竖头欠稳;立位:双下肢可支撑体重,扶持立位时竖头不稳;手抓位:无主动取物意识,留握差;异常姿势:竖头不稳;肌张力:双下肢肌张力增高;关节活动度:股角100°、腘窝角120°、足背屈角30°,围巾征不过中线;语言及智力评价:可逗笑,能笑出声,偶有“a”音,蒙面试验阴性。辅助检查:血清甲状腺功能指标、维生素D3及肝肾功能指标未见异常,血串联质谱及尿气相色谱—质谱未见异常。染色体核型分析:46,XX,未见结构异常。2016版儿心量表检测结果显示,大运动相当于2.5 月[发育商(DQ)33分],精细动作相当于1.0月(DQ 13分),适应能力相当于3.5 月(DQ 46 分),语言相当于3.0 月(DQ 40分)。结论:智龄2.6月,DQ 34分。表面肌电图结果示,被动状态下,双侧腘绳肌、背阔肌及右侧斜方肌肌张力稍增高,左侧斜方肌肌张力增高。双髋关节正位片未见明显异常。头颅MRI 未见明显异常。入院诊断:1 全面发育迟滞;2 遗传代谢性疾病?予关节松动、作业疗法及理疗等康复治疗,间断康复训练2个疗程,均因体质差继发感染而停止训练。
因患儿全面发育迟滞,经医学伦理审核及家属知情同意后,采集患儿及其父母外周血送基因检测。使用CASAVA1.8.2 软件将原始数据转化为可识别的碱基序列,获得靶向区域变异位点的信息。通过在线生物预测软件分析蛋白质损伤,获取变异的位点。测序分析发现,患儿ZC4H2 基因第2 外显子c.82G>A 杂合半合子变异,该变异导致该基因序列第82 位的碱基G 变异为A,导致该基因编码蛋白的第28 氨基酸位置上谷氨酸变异为赖氨酸(p. Glu28Lys),可能造成蛋白质功能遭受严重影响,该变异位点在美国医学遗传学与基因组学会数据库尚未见报道。该变异不归类为多态性变化,在人群中发生频率极低(所参考的数据库为1000Genomes 和dbSNP)。家系验证结果表明,父母均为该基因位点野生型,符合X 染色体隐性遗传规律(见图1)。
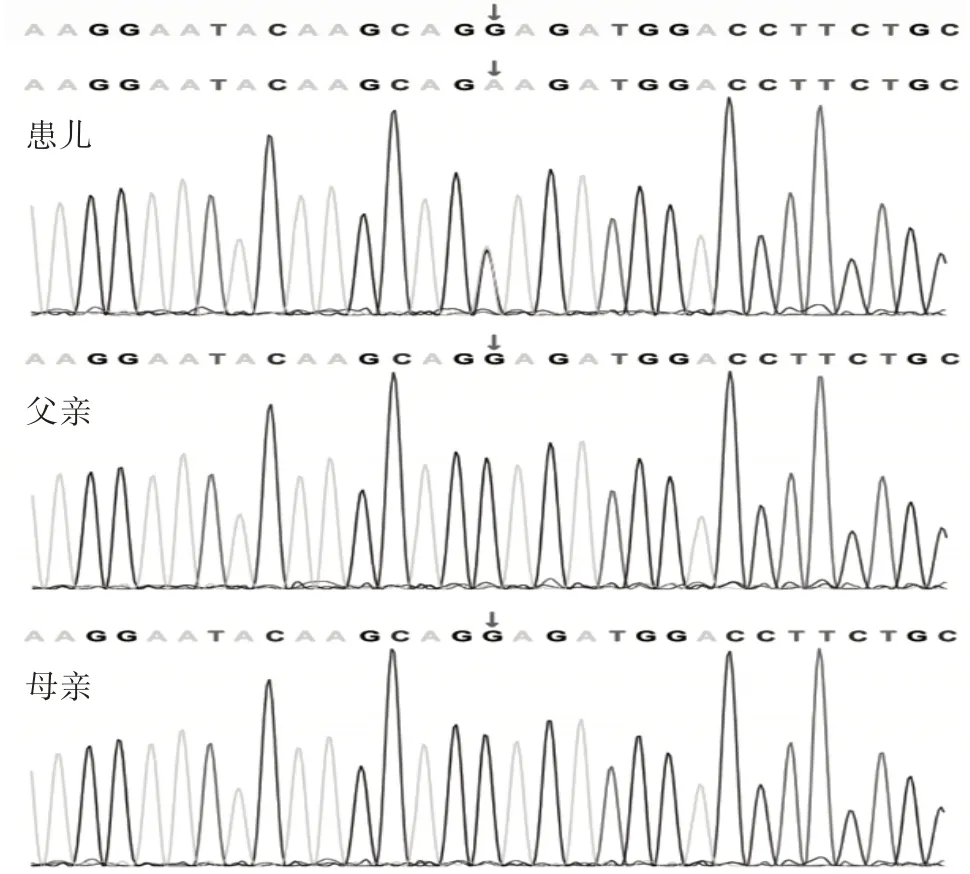
图1 患儿及其父母ZC4H2基因c.82G>A变异测序峰图
根据美国医学遗传学与基因组学会联合美国分子病理学会2015 年制订的“基因序列变异解释的标准和指南”进行致病性分析[7],ZC4H2 基因c.82G>A变异的致病性证据强度为“PM2+PM6+PP3”,为造成患儿发病临床意义不明性变异。但ZC4H2 基因c. 82G>A 变 异位 点在 SIFT、Polyphen2、Mutation-Taster 软件的预测结果分别为致病、可能致病及致病。
结合患儿双手掌指关节挛缩、智力障碍、发育迟滞症状和基因检测结果,诊断ZC4H2 基因c.82G>A变异所致的ZARD 明确,告知患儿家属该病目前尚无特效治疗,建议定期康复治疗。
2 讨论
ZC4H2 基因定位于Xq11.2,基因组全长约118.33 kb,包含5 个外显子和4 个内含子,编码的ZC4H2 蛋白共224 个氨基酸,包括1 个锌指结构域、4 个半胱氨酸残基、2 个组氨酸残基及1 个卷曲螺旋结构域。ZC4H2 基因在胚胎发育过程中和未成熟神经元中表达量最高,在出生后和成熟神经元中表达量均下降,表明其在神经系统发育中具有重要作用[2]。ZC4H2 基因表达于人体各器官,特别是胎儿大脑,可能与胎儿时期神经系统的发育密切相关。目前ZC4H2 基因变异的致病机制尚不清楚,但在斑马鱼模型中,ZC4H2 同源基因敲低或敲除可导致斑马鱼游动异常、运动神经元发育受损及GABA 能中间神经元显著减少[8]。MAY 等[9]研究发现,对斑马鱼ZC4H2 基因纯合敲除,表现出游动异常、抽搐增加、眼球运动缺陷和胸鳍挛缩,脊髓和大脑中GABA能中间神经元减少,而感觉和运动神经元表现正常,认为ZC4H2蛋白对大脑和脊髓GABA能中间神经元的正常发育至关重要。
目前已报道的ZC4H2 基因变异约为109 种,包括错义变异30 种、无义变异7 种、移码变异8 种、剪接位点突变2 种,位于非编码区RNA 和非翻译区的变异有 62 种(https://www. ncbi. nlm. nih. gov/clinvar)。ZC4H2 基因变异谱包括新生和复发遗传错义变异(多为男性患者受累)及新生变异、移码变异、无义变异和部分ZC4H2 基因缺失(多为女性患者受累)[10],最常见的报道为错义变异。目前,基因型—表型之间的相关性尚未明确。WANG 等[2]发现,携带相同变异的女性可能具有不同的临床表型,并认为非随机XCI 可能是这种现象的原因,但同时发现即使在携带相同变异和具有相似临床表型的女性患者中,其XCI 也不同。1 例因ZC4H2 基因杂合微缺失而表现为严重表型的女性患者和另外1 例因p.Q50X 无义变异而表现为轻度表型女性患者均显示随机XCI,提示XCI 与疾病表型之间并没有很强的相关性。也有研究认为,ZC4H2 基因的R18K、V63L、L66H、R198Q、P201S 及 R213W 这 6 种错义变异是导致半合子男性和杂合子女性患者AMC 合并X 连锁智力障碍的原因[11]。另外女性患者血液或皮肤成纤维细胞的XCI 模式从随机到偏斜不一,对预测其临床表型并无帮助,有报道1 例严重受累女性胎儿皮肤成纤维细胞的XCI 比率为61∶39,而其他轻到重度女性胎儿血液淋巴细胞的XCI 比率为100∶0 到 80∶20[12],而文献报道无症状携带者女性患者 XCI 比率大于 95∶5[13]。但最近一项研究表明,女性XCI 的机制不一定与疾病表型相关,在这方面,XCI 似乎与女性携带者温和表型相关[2]。本例患儿ZC4H2 基因c.82G>A 变异为错义变异,与文献报道一致。由于技术限制,本例患儿未能检测血液或皮肤成纤维细胞的XCI比率。
儿童和成年男女ZARD 患者的常见临床表型包括生后生长发育迟缓、全身肌张力减退、运动发育迟缓、行走障碍、痉挛、反射亢进、尿失禁、构音障碍—表达性语言缺陷、语言表达能力差或缺失、智力障碍、流涎、吞咽困难(包括口腔运动功能障碍的咀嚼困难、进食困难)、面瘫、高额头、高发际线、眼运动失用、斜视、朝天鼻、小后颌、AMC、肩部运动受限、肘及腕挛缩、掌指关节挛缩、任一手指桡偏、膝关节屈曲挛缩、马蹄内翻足畸形足、跟腱挛缩、远端肢体肌肉萎缩或无力、男性小阴茎或隐睾等[10]。在男性患者,ZARD 的表型多为智力障碍、伴智力障碍AMC、胎儿发育迟缓、生后生长迟缓、痉挛、四肢瘫痪、小头畸形和性腺功能减退;在女性患者,ZARD 的表型多为未受累的携带者、伴智力障碍AMC、生后生长迟缓、语言落后或缺失、痉挛、不能行走、远端肌肉萎缩、短颈伴转动受限、窄胸和肩膀活动受限[9]。但新发变异ZARD 女性患者临床表型按发生频率高低依次为不能行走(94%)、远端肌无力(90%)、运动发育迟缓(90%)、短颈伴转动受限(86%)、肩部运动受限(86%)、屈指畸形(84%)、轻微下颌后缩(82%)、智力障碍(80%)、髋关节挛缩(79%)、远端肌萎缩(76%)、斜视(75%)、尿失禁(75%)、窄胸及窄肩胸(73%)、语言表达匮乏(73%)、生后生长迟缓(71%)及痉挛状态(71%)[10]。本例患儿为女性,基因测序发现为新发变异,其临床症状包括生长发育迟滞、斜视、双手掌指关节挛缩、双手拇指手指尺偏及智力障碍,与文献报道一致,但该患儿双下肢肌张力增高,与文献报道不符,提示ZARD 临床表型有高度异质性。
有研究显示,由于X 染色体失活,与男性相比,女性患者症状较轻,脑部MRI 可有轻微异常[14]。本例患儿头颅MRI 未见异常,与文献报道不同。OKUBO 等[14]报道ZC4H2 基因变异(仅包括ZC4H2 基因的 395 kb 微缺失)致 AMC 伴脑萎缩、痉挛性四肢瘫痪和智力障碍的严重女性病例,但作者指出,进行性弥漫性脑萎缩及严重的身体和神经表现是否仅由ZC4H2 基因变异所致尚不清楚,需积累更多女性患者资料以建立基因型与表型的相关性。
ZARD 为遗传性疾病,目前治疗主要以对症为主。尽管尚无法明确ZC4H2 基因c.82G>A 变异位点的致病性,但患儿有ZARD 典型临床表现,且c.82G>A 变异不属于多态性变化,在人群中发生的频率极低,故综合考虑患儿基因检测结果及临床表型,可基本确认ZC4H2 基因c.82G>A 变异为患儿致病性变异。如需进一步明确ZC4H2 基因c. 82G>A变异位点的致病性,仍需要进行大样本的回归分析并行转基因动物的模型验证。
