高比活度碳-14标记氟噻草胺的合成与分析
2022-09-21王国通宋明钰杨征敏
王国通 宋明钰 杨征敏 周 兵
(上海启甄同位素标记合成研究中心,上海 201403)
氟噻草胺(1,英文名:flufenacet,系统名:N-(4-氟苯基)-N-异丙基-2-((5-(三氟甲基)-1,3,4-噻二唑-2-基)氧基)乙酰胺,图1)是德国拜耳公司创制的芳氧乙酰胺类除草剂[1],可抑制植物的细胞分裂和生长[2],主要用于玉米、大豆、番茄、马铃薯和水稻等作物田间的一年生禾本科杂草、莎草及一些小粒阔叶杂草的防控,具有高效、低毒、安全性高等优点,可与多种除草剂复配扩大杀草谱[3]。氟噻草胺市场主要分布在欧洲、北美和东南亚部分地区。当前氟噻草胺在我国的专利保护到期,因其独特的作用方式和优良的复配特性,国内科研单位及农药企业陆续开展了针对该除草剂的研发工作和登记代谢试验[4-8]。
根据我国2017年9月颁布的《农药登记资料要求》规定,放射性同位素碳-14标记物是氟噻草胺在农作物、环境和动物中登记代谢试验所必需的放射性示踪剂[9-11]。文献报道了两种碳-14标记氟噻草胺[12-13],但未见有关其合成与分析的报道。为满足我国氟噻草胺登记代谢试验对放射性示踪剂的要求,在综合考虑该药物的化学稳定性、代谢稳定性和毒理学重要性等因素后,本研究选择氟噻草胺分子中具有芳香性的苯环和噻二唑环为标记单元,以标记单元中的碳为标记位点(2和3,图1)。借助标记物(2和3)开展的登记代谢试验,证实这两种标记物作为放射性示踪剂满足我国农药登记代谢试验的要求。综上,本文对这两种碳-14标记氟噻草胺进行合成与分析(合成路线见图2、3),以期为氟噻草胺的同位素示踪研究提供参考。
1 材料与方法
1.1 材料与试剂

图1 氟噻草胺(1)及其放射性同位素碳-14标记物(2,3)(*表示碳-14标记位点)Fig.1 Flufenacet (1) and its radioisotope carbon-14 labelled counterparts (2,3)(* indicates carbon-14 labelled sites)
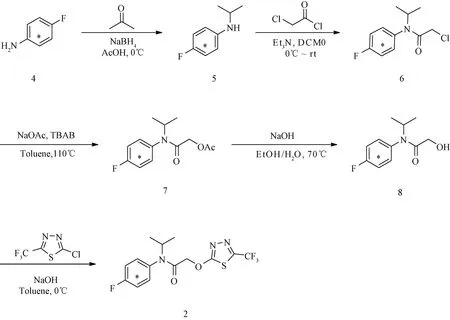
图2 苯环碳-14标记氟噻草胺(2)的合成路线(*表示碳-14标记位点)Fig.2 Synthetic route of [phenyl-U-14C]flufenacet (2)(* indicates carbon-14 labelled sites)

图3 [噻二唑环-2-14C]氟噻草胺(3)的合成路线(*表示碳-14标记位点)Fig.3 Synthetic route of [thiadiazole-2-14C]flufenacet (3)(* indicates carbon-14 labelled sites)
同位素原料4-氟[U-14C]苯胺(比活度2 057.20 MBq·mmol-1,放化纯度和化学纯度均大于98%)和[14C]硫氰酸钠(比活度2 049.80 MBq·mmol-1,放化纯度和化学纯度均大于98%)均为自行制备;闪烁液Optiphase HiSafe 3,购于美国PE公司;高效液相色谱(high performance liquid chromatography,HPLC)和高效液相色谱-质谱(high performance liquid chromatography-mass spectrometry,HPLC-MS)联用技术所用甲醇分别为色谱级和质谱级,购于美国Fisher Scientific公司;Milli-Q Advantage A10超纯水制备仪,购于默克化工技术(上海)有限公司;其他试剂均为市售分析纯,溶剂按照文献[14]的方法纯化。
1.2 目标物N-(4-氟[U-14C]苯基)-N-异丙基-2-((5-(三氟甲基)-1,3,4-噻二唑-2-基)氧基)乙酰胺(2,简称为苯环碳-14标记氟噻草胺)的制备
1.2.1 中间体4-氟-N-异丙基[U-14C]苯胺(5)的合成 在0~5℃下,将4-氟[U-14C]苯胺 (4,28.3 mg,514.30 MBq)、丙酮(21.8 mg)与乙酸(2 mL)混合,搅拌20 min后加入NaBH4(19 mg)[15],继续搅拌。在线放射性高效液相色谱-二极管阵列检测器串联质谱(HPLC-FSA/PDA/MS,简称HFPM:Waters Alliance e2695 HPLC-AIM ν.ARC FSA/Waters 2998 PDA/Waters Acquity QDa MS多检测器联用系统与HPLC-PDA/MS系统购于美国沃特世公司,FSA购于美国AIM Research公司)监测显示,反应1 h原料(4)消耗完毕。以饱和碳酸氢钠溶液调节反应液pH值至8,经乙酸乙酯(ethyl acetate,EA)萃取(30 mL×5)、饱和食盐水洗涤、无水硫酸钠干燥和减压脱溶得黄色液体4-氟-N-异丙基[U-14C]苯胺(5,32.9 mg,437.34 MBq,85%)。放射性薄层成像(Radio-TLC)分析:比移值(retention factor value)Rf(5)=0.65;展开剂:V(EA)/V(PE)=1/10(体积比,下同),PE为石油醚(petroleum ether)。色谱条件:Diamonsil C18色谱柱(5 μm,4.6 mm × 150 mm,美国迪马公司);流速1.00 mL·min-1;波长254 nm;柱温30℃;进样量10 μL;梯度洗脱(min/%A)控制:0/20,1/20,10/100,13/100,14/20,15/20。A为甲醇,B为含0.05%甲酸的水溶液。下文如无特殊说明,HPLC分析均使用此条件。
1.2.2 中间体2-氯-N-(4-氟[U-14C]苯基)-N-异丙基乙酰胺(6)的合成 在氩气保护和冰-水浴冷却下,将氯乙酰氯(36.5 mg)滴入4-氟-N-异丙基[U-14C]苯胺(5,32.9 mg,437.30 MBq)和三乙胺(42.9 mg)的无水二氯甲烷(dichloromethane,DCM,5 mL) 溶液中,滴加完毕撤去冰-水浴,自然升至室温[16]。HFPM监测显示,室温搅拌0.5 h原料(5)消耗完毕。以水(10 mL )淬灭反应,反应混合液经DCM(30 mL×5)萃取、饱和食盐水洗涤、无水硫酸钠干燥、减压脱溶和快速硅胶柱层析[V(EA)/V(PE)=1/3]纯化得黄色液体2-氯-N-(4-氟[U-14C]苯基)-N-异丙基乙酰胺(6,46.5 mg,415.14 MBq,95%)。Radio-TLC分析:Rf(6)=0.54;展开剂:V(EA)/V(PE)=1/5。产物(6)在HFPM中的保留时间为7.734 min。梯度洗脱(min/%A)控制:0/50,1/50,10/100,12/100,13/50,15/50;其余条件同1.2.1。
1.2.3 中间体2-((4-氟[U-14C]苯基)(异丙基)氨基)-2-氧乙基乙酸酯(7)的合成 在氩气保护和室温下,将2-氯-N-(4-氟[U-14C]苯基)-N-异丙基乙酰胺 (6,46.5 mg,415.14 MBq)、NaOAc(32.5 mg)和四丁基溴化铵(缩写为TBAB,6.5 mg)溶于甲苯(2 mL),升温至110℃,继续搅拌[17]。HFPM监测显示,反应1 h原料(6)消耗完毕。体系降至室温,加水(10 mL )淬灭反应,反应混合物经EA(20 mL×5)萃取、饱和食盐水洗涤、无水硫酸钠干燥和减压脱溶得黄色液体2-((4-氟[U-14C]苯基)(异丙基)氨基)-2-氧乙基乙酸酯 (7,45.9 mg,373.70 MBq,90%)。Radio-TLC分析:Rf(7) = 0.65;展开剂:V(EA)/V(PE) = 1/6,重复展开2次。产物(7)在HFPM中的保留时间为7.084 min。色谱条件同1.2.2。
1.2.4 中间体N-(4-氟[U-14C]苯基)-2-羟基-N-异丙基乙酰胺(8)的合成 在氩气保护和室温下,将2-((4-氟[U-14C]苯基)(异丙基)氨基)-2-氧乙基乙酸酯 (7,45.9 mg,373.70 MBq)、NaOH(14.5 mg)、乙醇(0.5 mL)和水(0.5 mL)混合,升温至70℃,继续搅拌[18]。HFPM监测显示,反应1 h原料(7)消耗完毕。体系降至室温,减压脱除乙醇,加水(5 mL)稀释,混合液经DCM(20 mL×5)萃取、无水硫酸钠干燥和减压脱溶得黄色固体N-(4-氟[U-14C]苯基)-2-羟基-N-异丙基乙酰胺 (8,38.5 mg,336.33 MBq,90%)。Radio-TLC分析:Rf(8)=0.55;展开剂:V(EA)/V(PE) =1/3,重复展开2次。产物(8)在HFPM中的保留时间为5.977 min。色谱条件同1.2.2。
1.2.5 苯环碳-14标记氟噻草胺(2)的合成 在氩气保护和冰-水浴冷却下,将2-氯-5-(三氟甲基)-1,3,4-噻二唑(61 mg)滴入N-(4-氟[U-14C]苯基)-2-羟基-N-异丙基乙酰胺 (8,38.5 mg,336.33 MBq)、NaOH(500 mg)、甲苯(4 mL)与水(0.5 mL)的混合液中,滴加完毕继续搅拌[3]。HFPM监测显示,反应1 h原料(8)消耗完毕。加水(5 mL)稀释,混合液经EA(20 mL×5)萃取、饱和食盐水洗涤、无水硫酸钠干燥和快速硅胶柱层析[V(EA)/V(PE)=1/8]纯化得放射性白色固体(2,35.45 mg,201.65 MBq,60%)。Radio-TLC分析:Rf(2)=0.50;展开剂:V(EA)/V(PE)=1/3, 连续展开3次。产物(2)在HFPM中的保留时间8.234 min。梯度洗脱(min/%A)控制:0/60,1/60,10/100,11/100,13/60,15/60;其余条件同1.2.1。
1.3 目标物N-(4-氟苯基)-N-异丙基-2-((5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-基)氧基)乙酰胺(3,简称为噻二唑环碳-14标记氟噻草胺)的制备
1.3.1 中间体2-(丙-2-亚烷基)肼基-1-[14C]碳硫酰胺(10)的合成 在氩气保护和室温下,将[14C]硫氰酸钠(9,83 mg,2 049.80 MBq)、硫酸肼(130 mg)溶于水(5 mL)和丙酮(0.1 mL)混合液,升温至110℃,继续搅拌。HFPM监测显示,搅拌4 h原料(9)消耗完毕。体系降至室温,静置2 h,于4℃下过夜析晶,获得白色晶体2-(丙-2-亚烷基)肼基-1-[14C]碳硫酰胺 (10,66 mg,1 024.90 MBq,50%)。产物(10)在HFPM中的保留时间为5.023 min。梯度洗脱(min/%A)控制:0/20,1/20,10/60,13/60,15/20;其余色谱条件同1.2.2。
1.3.2 中间体氨基[14C]硫脲(11)的合成 在氩气保护和室温下,将2-(丙-2-亚烷基)肼基-1-[14C]碳硫酰胺(10,66 mg,1 024.90 MBq)的水(5 mL)溶液在搅拌下升温至115℃。HFPM 监测显示,继续搅拌5 h原料(10)消耗完毕。减压脱水得白色固体氨基[14C] 硫脲 (11,28 mg,614.94 MBq,60%),直接进行下一步。
1.3.3 中间体5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-胺(12)的合成 在氩气保护和室温下,将三氟乙酸(0.08 mL,1.05 mmol)、三氯氧磷(0.2 mL) 依次滴入搅拌的氨基[14C]硫脲(11,28 mg,614.94 MBq)的甲苯(2.5 mL)溶液,升温至80℃。HFPM监测显示,继续搅拌5 h原料(11)消耗完毕。体系降至室温,以冰水淬灭反应;以20% NaOH溶液将体系pH值调至8,经乙酸乙酯(20 mL×5)萃取、无水硫酸钠干燥、减压脱溶和快速硅胶柱层析(V(EA)/V(PE)=1/2)纯化得白色固体5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-胺(12,46 mg,553.52 MBq,90%)。Radio-TLC分析:Rf(12)=0.45;展开剂:V(EA)/V(PE) = 1/2。产物(12)在HFPM中的保留时间为4.101 min。
1.3.4 中间体2-溴-5-三氟甲基-1,3,4-[2-14C]噻二唑(13)的合成 在氩气保护和室温下,将5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-胺(12,50 mg,553.52 MBq)、溴化铜(80 mg)和亚硝酸叔丁酯(50 mg)与乙腈溶液(4 mL)混合,搅拌,升温至65℃。HFPM监测显示,反应3 h原料(12)消耗完毕[19]。体系降至室温,以15%盐酸(15 mL)淬灭反应,反应混合物经甲苯(20 mL×5)萃取、无水硫酸钠干燥和减压脱溶得淡黄色液体2-溴-5-三氟甲基-1,3,4-[2-14C]噻二唑 (13,50.5 mg,442.89 MBq)。Radio-TLC分析:Rf(13) =0.60;展开剂:V(EA)/V(PE) =1/3。产物(13)在HFPM中的保留时间为6.291 min。
1.3.5 目标物噻二唑环碳-14标记氟噻草胺(3)的合成 在氩气保护和冰-水浴冷却下,将2-溴-5-三氟甲基-1,3,4-[2-14C]噻二唑 (13,50.5 mg,442.89 MBq)滴入N-(4-氟苯基)-2-羟基-N-异丙基乙酰胺(14,68 mg)和NaOH(500 mg)的甲苯(4 mL)和水(0.5 mL)混合液,滴加完毕后维持此温度搅拌[3]。HFPM监测显示,继续搅拌1 h原料(3)消耗完毕。向反应混合物加水(5 mL)稀释,经EA(20 mL×5)萃取、饱和食盐水(10 mL×3)洗涤、无水硫酸钠干燥和快速硅胶柱层析(V(EA)/V(PE) =1/8)纯化得白色固体 (3,51 mg,287.86 MBq)。Radio-TLC分析:Rf(3) = 0.50;展开剂:V(EA)/V(PE) =1/3,连续展开3次。产物(3)在HFPM中的保留时间为8.241 min。色谱条件同1.2.5。
1.4 同位素标记物的质量指标分析
同位素标记物(2)和(3)的质量指标主要包括化学纯度、放化纯度和比活度,具体分析测定方法如下。
1.4.1 化学纯度的测定 将标记物(2)和(3)分别配置浓度为4、6、8 μg·mL-1的甲醇标准溶液,参照文献[20-21]以外标法测定其化学纯度,HPLC色谱条件:XBridge C18 色谱柱(5 μm,4.6 mm × 250 mm,美国迪马公司);流速1 mL·min-1;波长254 nm;柱温30℃;进样量10 μL;梯度洗脱(min/%A)控制:0/40,1/40,45/100,50/100,55/40,60/40;A为甲醇,B为含0.05%甲酸的水溶液。
1.4.2 放化纯度的测定 采用放射性薄层成像分析法(TLC-IIA)(Eckert & Ziegler AR-2000薄层放射性扫描仪,德国Eckert & Ziegler公司)和Waters Alliance e2695 HPLC-AIM ν.ARC FSA在线放射性高效液相色谱(HPLC-FSA)分别测定标记物(2)和(3)的放化纯度[22-23]。
TLC-IIA分析:标记物(2)、(3)和氟噻草胺标样在同一硅胶层析薄板(5 mm×20 mm)上展开,其比移值(Rf)均为0.67。TLC条件:V(EA)/V(PE)=1/3,连续展开4次。
HPLC-FSA分析:色谱条件同1.4.1;PerkinElmer Tri-Carb 4910TR流动液体闪烁测量仪(美国PE公司),使用Optiphase HiSafe 3闪烁液,其流速为8.00 mL·min-1。标记物(2)和(3)在HPLC-FSA色谱图中均显示1个具有放射性特征的规则色谱峰。
1.4.3 比活度的测定 用微量注射器分别精确移取标记物(2)和(3)的溶液(10 μL,0.01 μg·mL-1)各6份,用液体闪烁测量仪测量其放射性活度,根据测量结果计算比活度[22-23]。
2 结果与分析
本研究以4-氟-[U-14C]苯胺和[14C]硫氰酸钠为同位素原料,分别经五步放化反应制备了放射性同位素碳-14标记物(2)和(3),其放化总收率分别为39%和14%。通过1H NMR (Varian 400 MHz 核磁共振仪,以TMS为内标)和ESI-MS (Waters Alliance e2695 HPLC-Acquity Qda MS/Waters 2998 PDA)分别对各产物进行表征,表征数据如下:
4-氟-N-异丙基[U-14C]苯胺(5):1H NMR (400 MHz,DMSO-d6),δ: 6.92 - 6.83 (m,2 H,Ar-H),6.51 (ddd,J= 6.7,5.3,2.9 Hz,2 H,Ar-H),5.22 (d,J= 8.1 Hz,1 H,NH),3.52 - 3.39 (m,1 H,CH),1.09 (d,J= 6.3 Hz,6 H,CH3)。ESI-MSm/z: 156[M+H]+。
2-氯-N-(4-氟[U-14C]苯基)-N-异丙基乙酰胺(6):1H NMR (400 MHz,DMSO-d6),δ: 7.40 - 7.23 (m,4 H,Ar-H),4.79 - 4.68 (m,1 H,CH),3.79 (s,2 H,CH2),0.96 (t,J= 8.4 Hz,6 H,CH3)。ESI-MSm/z: 232[M+H]+,254[M+Na]+。
2-((4-氟[U-14C]苯基)(异丙基)氨基)-2-氧乙基乙酸酯(7):1H NMR (400 MHz,CDCl3),δ:7.19-7.11 (m,4 H),4.94 (tt,J= 6.8,6.8 Hz,1 H),4.18 (s,2 H),2.12 (s,3 H),1.05 (d,J= 6.8Hz,6 H)。ESI-MSm/z: 256[M+H]+。
N-(4-氟[U-14C]苯基)-2-羟基-N-异丙基乙酰胺(8):1H NMR (400 MHz,DMSO-d6),δ: 7.33 - 7.20 (m,4 H,Ar-H),4.77 (dt,J= 13.4,6.6 Hz,1 H,CH),4.45 (t,J= 5.7 Hz,1 H,OH),3.47 (d,J= 5.6 Hz,2 H,CH2),0.95 (d,J= 6.8 Hz,6 H,CH3)。ESI-MSm/z: 214[M+H]+,236[M+Na]+。
目标物苯环碳-14标记氟噻草胺(2):1H NMR (400 MHz,DMSO-d6),δ: 7.41 (dd,J= 8.6,5.2 Hz,2 H,Ar-H),7.35 (t,J= 8.6 Hz,2 H,Ar-H),4.77 (s,2 H,CH2),4.72 (dd,J= 13.5,6.8 Hz,1 H,CH),0.98 (d,J= 6.8 Hz,6 H,CH3)。19F NMR (76 MHz,CDCl3) δ: -58.36 (s,3F),-108.74 ~ -109.08 (m,1F)。ESI-MSm/z: 196,388 [M+Na+2]+。
2-(丙-2-亚烷基)肼-1-[14C]碳硫酰胺(10):1H NMR (400 MHz,DMSO-d6),δ: 9.89 (s,1 H,NH),7.98 (s,1 H,NH2),7.50 (s,1 H,NH2),1.91 (s,3 H,CH3),1.90 (s,3 H,CH3)。ESI-MSm/z: 134[M+Na]+。
5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-胺(12):1H NMR (400 MHz,DMSO-d6) δ:8.03 (s,2 H);19F NMR (76 MHz,DMSO-d6) δ:-56.50 (s,3F)。ESI-MSm/z: 172[M+H]+。
2-溴-5-三氟甲基-1,3,4-[2-14C]噻二唑(13):19F NMR (76 MHz,DMSO-d6) δ: -56.64 (s,3F)。EI-MSm/z: 234[M+2]+,236[M+2+2]+。
目标物噻二唑环碳-14标记氟噻草胺(3):1H NMR (400 MHz,CDCl3) δ: 7.40 (dd,J= 8.6,5.1 Hz,2 H,Ar-H),7.38 (t,J= 8.6 Hz,2 H,Ar-H),4.77 (s,2 H,CH2),4.72 (dd,J= 13.5,6.8 Hz,1 H,CH),0.99 (d,J= 6.8 Hz,6 H,CH3)。19F NMR (76 MHz,CDCl3) δ: -58.36 (s,3F),-108.74 ~ -109.08 (m,1F)。ESI-MSm/z: 194,388[M+Na+2]+。
TLC-IIA分析显示,标记物(2)和(3)经展开后在硅胶薄板上均显示1个具有放射性特征的斑点,且该斑点形状和比移值与同一硅胶板上同时展开的氟噻草胺标样(UV显色)一致,表明标记物(2)和(3)中均仅含有1种放射性碳-14标记氟噻草胺。HPLC-FSA分析显示,标记物(2)和(3)的放化纯度均大于98%(图4),与TLC-IIA分析结果一致。HPLC外标法定量分析显示,标记物(2)和(3)的化学纯度均大于98%。根据LSC分析结果计算可知,两种标记物的比活度分别为2 049.80 MBq·mmol-1和2 042.40 MBq·mmol-1,其总活度分别为201.65 MBq和287.86 MBq。
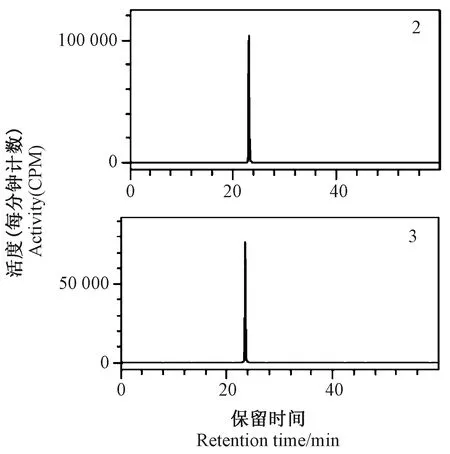
图4 两种碳-14标记氟噻草胺(2,3)的在线放射性高效液相色谱图Fig.4 Online radio-chromatogam (HPLC-LSC) of carbon-14 labelled flufenacet (2,3)
在核磁共振氢谱中,标记物(2)和(3)上氢原子的化学位移、峰型和耦合常数与氟噻草胺标样基本一致[24-25]。氟噻草胺(1)的精确相对分子质量为363[M],其标样质谱图中典型碎片离子峰和加合离子峰对应的质核比分别为194和386[363+Na]+。标记物(2)质谱图(图5)中典型碎片离子峰和加合离子峰对应的质核比(m/z)分别为196[194+2]和388[M+Na+2]+,其中m/z196表明标记核素碳-14处于氟噻草胺的典型离子碎片中,加合离子m/z388与363[M]的差值进一步验证了标记物(2)中含有碳-14。标记物(3)质谱图(图5)中典型碎片离子峰和加合离子峰对应的质核比(m/z)分别为194(与氟噻草胺典型离子碎片质核比一致,其中不含碳-14)和388[M+Na+2]+,其中两种加合离子m/z388与363[M]的差值进一步印证了标记物(3)中存在碳-14。标记物(2)和(3)在HPLC中的保留时间均为26.355 min,与氟噻草胺标样在相同色谱条件下的保留时间一致。结合标记物的合成路线、氢谱数据、质谱特征、HPLC保留时间和物质放射性特性,可以确定标记物(2)和(3)分别为N-(4-氟[U-14C]苯基)-N-异丙基-2-((5-(三氟甲基)-1,3,4-噻二唑-2-基)氧基)乙酰胺和N-(4-氟-苯基)-N-异丙基-2-((5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-基)氧基)乙酰胺。
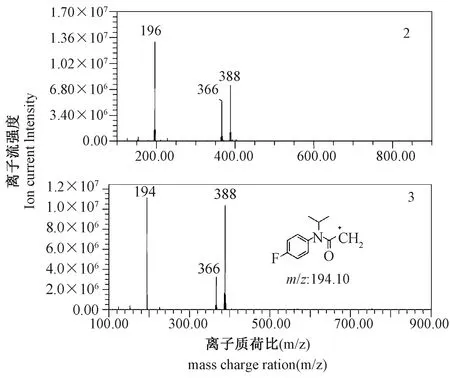
图5 两种碳-14标记氟噻草胺(2和3)的电喷雾电离-质谱图Fig.5 Mass spectrum of two carbon-14 labelled counterparts (2 and 3) of flufenacet (ESI-MS)
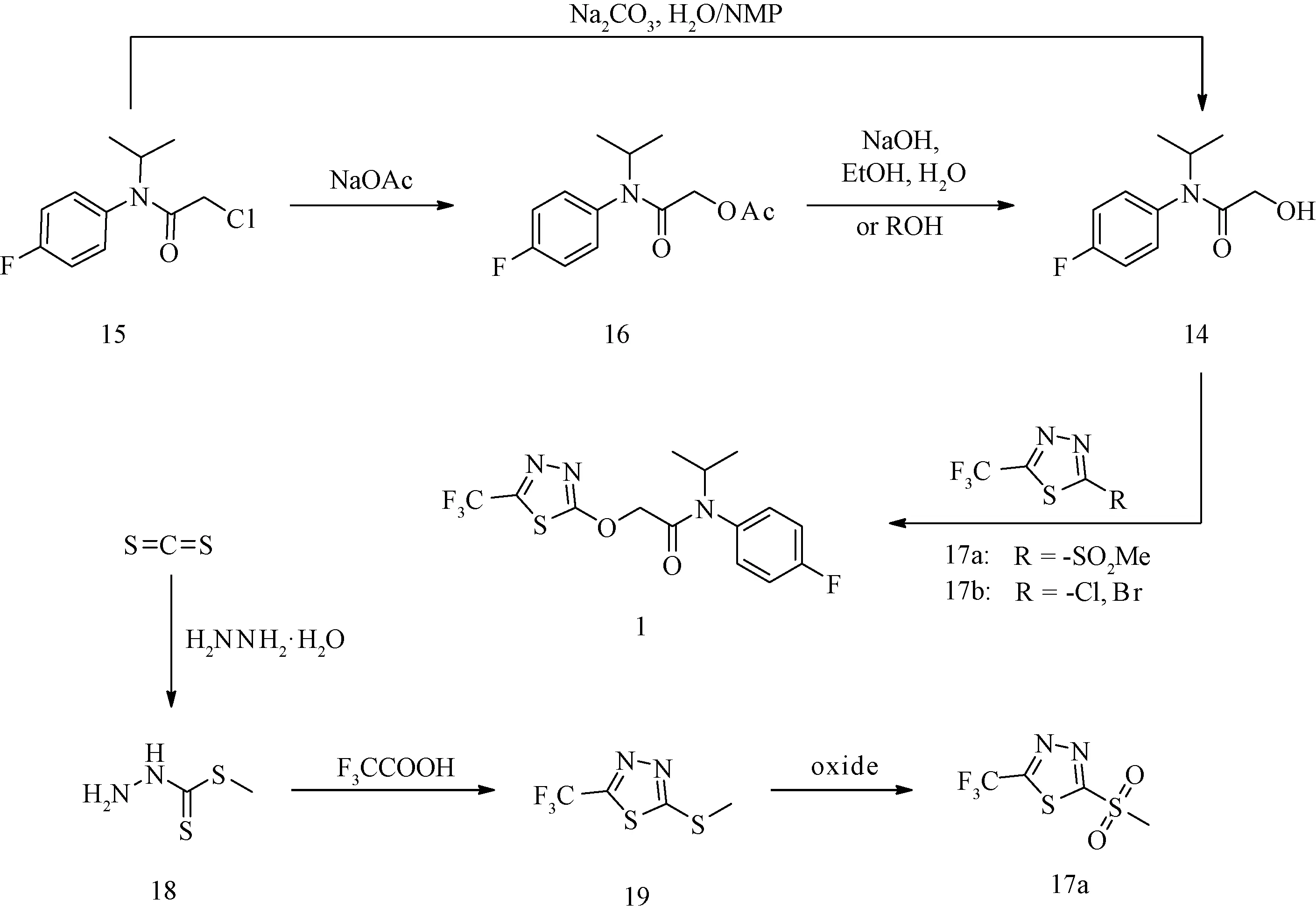
图6 氟噻草胺(1)的常见合成技术路线汇总Fig.6 Summary of common synthetic routes of flufenacet (1)
3 讨论
在农药碳-14标记合成时,标记位点的选择是首要考虑的问题。按照我国现行法规要求,碳-14标记通常要求对农药分子的碳骨架进行标记,且优先考虑对碳环标记[9-11]。本研究以氟噻草胺分子中具有芳香性的苯环和噻二唑环为标记单元,从标记的可行性、难易程度和成本等角度选取标记单元中适当的碳为标记位点,相应的两种标记物(2和3)中碳-14标记牢固而不易脱落,可满足我国农药登记代谢试验对标记物的要求。
在合成标记物(2)的模拟冷反应(图6)中,2-氯-N-(4-氟苯基)-N-异丙基乙酰胺(15)与碳酸钠在水与N-甲基吡咯烷酮(缩写为NMP)混合液中加热一步反应可获得中间体N-(4-氟苯基)-2-羟基-N-异丙基乙酰胺(14)[26];中间体(15)与醋酸钠发生酯化反应,所得酯(16)经碱性水解或者酯交换也可获得中间体(14)[27]。相比较而言,前法通过一步反应可获得中间体(14),看似为最理想的标记方法,但试验证实该法所得微量液态产物(14)中夹杂的超微量高沸点溶剂NMP难以除去而引起后续反应收率降低,甚至反应失败;尽管后法经两步反应获得中间体(14),但这两步的反应条件温和,后处理简单,反应收率近乎定量,因而标记合成中标记中间体(6)经两步反应来制备(8)。中间体(8)通过与2倍量的2-氯-5-(三氟甲基)-1,3,4-噻二唑反应而完全转化。
在合成标记物(3)的模拟冷反应(图6)中,噻二唑类化合物通常经肼基二硫代甲酸甲酯(18)或氨基硫脲与三氟乙酸关环制备。二硫化碳和三氟乙酸均为低沸点易挥发物质,使用这两种物质对应的含碳-14合成砌块制备噻二唑均存在因挥发而引起内照射的潜在风险,因而本研究放弃了经过(18)制备2-甲硫基-5-三氟甲基-1,3,4-噻二唑(19),以双氧水或过硫酸盐氧化为2-甲砜基-5三氟甲基-1,3,4-噻二唑(17a)的技术路线[28],而以无机盐[14C]硫氰酸钠(9)为起始同位素原料,后续制备的含碳-14中间体(10~12)均为固体,中间体(13)为难挥发物质,与这些含碳-14中间体反应所涉及的挥发性物质均为非放射性原料,因而在标记合成过程中不存在因放射性物质挥发而带来内照射的风险。氨基[14C]硫脲(11)与三氟乙酸、三氯氧磷反应关环获得5-(三氟甲基)-1,3,4-[2-14C]噻二唑-2-胺(12)。为了提高(12)的转化率,本研究通过经亚硝酸叔丁酯重氮化反应使中间体(12)转化为活性更高的噻二唑溴代物2-溴-5-三氟甲基-1,3,4-[2-14C]噻二唑(13);在碱性条件下,中间体(13)与1.5倍量的(14)反应可获得目标物(3)[29]。
4 结论
本研究以4-氟[U-14C]苯胺和[14C]硫氰酸钠为起始同位素原料,通过多步放射合成反应分别制备了苯环碳-14标记氟噻草胺(2)和噻二唑环中2-位碳-14标记氟噻草胺(3),其化学纯度和放化纯度均大于98%,比活度分别为2 049.80 MBq·mmol-1和2 042.40 MBq·mmol-1,总活度分别为201.65 MBq和287.86 MBq。两种碳-14标记氟噻草胺均可作为放射性示踪剂,用于氟噻草胺的同位素示踪研究,包括该除草剂在我国的登记代谢试验。
