一个MYO15A基因新的复合杂合位点突变导致先天性聋病例分析亓文晶1,2
2022-09-20王雅宁于萌萌张楠张巍古林涛
王雅宁 于萌萌 张楠 张巍 古林涛
1 山东第一医科大学(山东省医学科学院)(济南 250000);
2 山东第一医科大学第一附属医院(山东省千佛山医院)耳鼻喉科;
3 山东省济宁市邹城市兖矿新里程总医院; 4 广州嘉检医学检测有限公司
先天性聋是人类最常见的出生缺陷之一,在新生儿中的发病率可达1‰~3‰[1]。遗传因素是引起先天性聋的最主要原因,根据我国聋病分子流行病学调查显示,常见的耳聋相关基因主要有GJB2、SLC26A4、mtDNA12SrRNA及GJB3[2-4]。常染色体隐性非综合征型聋(autosomal recessive nonsyndromic hearing loss, ARNSHL)在非综合征型聋中约占80%,常见基因为:GJB2、SLC26A4、MYO15A、OTOF及CDH23[5]。
MYO15A基因自1995年发现至今已有200多个突变位点被报道并已证实与耳聋有关。本研究通过应用高通量测序(高精度临床外显测序)对一个耳聋家系样本进行测序和分析,发现该家系的致聋突变为MYO15A基因上C.3756+1G>A和C.4519C>T(p.R1507)构成的复合杂合突变,其中C.4519C>T(p.R1507)突变为国内首次报道的MYO15A基因新的致病位点,报告如下。
1 资料与方法
1.1研究对象 研究对象为山东省千佛山医院耳鼻咽喉科确诊的1个常染色体隐性遗传性聋核心小家系,包括1名子代双侧极重度聋患者(2岁,女),2名表型正常双亲(母亲24岁,父亲26岁)。子代患者基本信息和体格检查信息完整,听力检测、前庭功能检查、智力检查、颞骨CT及颅脑、内听道MRI及耳蜗水成像等影像学检查结果完整,且都已签署伦理知情同意书。
1.2研究方法
1.2.1高通量测序(高精度临床外显PLUS,由广州嘉检医学检测有限公司完成) 抽取该家系3人的外周静脉血3~5 ml,以盐析法提取基因组DNA,在Sim-100测定DNA浓度,经质检合格后,进行上机测序。使用高精度医学外显检测试剂盒,结合Illumuna Nextseq 500(Illumuna,加利福尼亚,美国)的高通量测序技术,对3份家系DNA样本进行5 177个OMIM上已知明确的致病基因的编码区和已知致病的内含子进行捕获测序,详细步骤如下:①文库构建:经Pre-Capture LM-PCR对纯化后文库进行扩增。等量混合文库后,加入人类外显子文库进行杂交,捕获外显子区域,再通过 PCR扩增富集捕获后产物。扩增后进行上机测序,测序平台为Illumuna Nextseq 500,读长模式为单端测序150 bp,每个样本的平均测序深度至少为224 x。②测序后获得的原始数据由Illumina basecalling Software 1.7进行处理。③有效数据通过BWA (version 0.7.5, http://bio-bwa.sourceforge.net/bwa.shtml)在线软件与人类参考基因组(genome GRCh37/hg19)对比得到目标基因组序列,得到BAM格式的最初对比结果。BAM 文件再利用在线软件Picard (version 1.96, https://sourceforge.net/projects/picard/files/picard-tools/1.96/)、GATK (version3.1-1, https://wiki.rc.usf.edu/index.php/Genome_Analysis_ToolKit_(GATK))与SAMtoolS (version0.1.18)进行去重复、局部重对比和碱基质量校正等处理。④使用GATK Haplotype Caller程序在目标区域找出位点的基因型。使用GATK Variant Filtration过滤突变。
1.2.2高通量测序结果分析 测序结果经生物信息学方法进行数据分析,对变异数据进行质控分析,检索嘉检内部数据库(http://opengk.com/)、dbSNP(单核苷酸多态性数据库)、ESP6500和ExAC(外显子组整合数据库)等人群数据库,标注单核苷酸多态性和低频良性变异。应用预测软件对变异的保守性、致病性和危害性进行预测。检索HGMD(人类基因突变数据库)、PubMed(美国国家生物信息中心开发的基于WEB的生物医学信息检索系统)、ClinVar(NCBI主办的与疾病相关的人类基因组变异数据库)等数据库,检索变异相关文献,分析文献内容,参考美国医学遗传学和基因组学学会(ACMG)变异分类指南,对变异进行分类,变异类型包括:错义、无义、同义、移码、整码、剪切等,筛选出最为可能的致病基因和突变进行验证。
1.2.3Sanger测序验证 通过UCSC网站查找上述候选阳性基因突变位点上下游序列,使用primer5设计特异性引物进行PCR扩增,再通过琼脂糖凝胶电泳法进行分析,然后将扩增产物进行Sanger测序得到基因片段序列,最后将测序结果与参考基因组序列进行比对,判断该位点的具体突变情况。
2 结果
2.1家系临床表现和遗传特征分析 该家系为一个两代常染色体隐性遗传性聋的3人核心小家系,即两名正常听力的父母及1名2岁患儿。患儿出生时听力筛查未通过,6月龄时复筛仍未通过,体格检查及智力发育正常。影像学检查示:颞骨CT、颅脑及内听道MRI及耳蜗水成像无明显异常,听力学检查示:鼓室导抗图为A型,听性脑干反应(ABR)、听觉稳态反应(ASSR)显示为双侧极重度感音神经性聋,双耳畸变产物耳声发射(DPOAE)各频率未引出。复习患儿病史及进行体格检查后排除了环境因素致聋的原因。
2.2高通量测序结果分析及突变基因变异位点验证 对患者样本进行高精度临床外显测序,综合测序质量和生物学信息分析,结合临床症状发现MYO15A基因上发生2个复合杂合突变。为验证测序结果,对家系中3个样本进行Sanger测序验证(图1),发现患者携带MYO15A的复合杂合突变C.3756+1G>A和C.4519C>T (p.R1507*);C.3756+1G>A这个剪切变异来源于父亲,位于mRNA剪接区域,序列高度保守,并且多种计算辅助算法预测这个变异可能会影响蛋白功能。C.4519C>T(p.R1507*)无义变异来源于母亲,该突变位于蛋白质高度保守的区域,可能会导致蛋白质合成提前出现氨基酸的终止密码。母亲携带杂合无义突变C.4519C>T(p.R1507*),父亲携带杂合剪切突变 3756+1G>A,这两个变异在参考人群基因数据库中频率较低。结合送检者的临床表现和家系分析,依据美国ACMG变异分类指南[6,7],这两个变异为1类-致病突变,证据分别都为1个非常强证据PVS1(pathogenic very strong ),2个中等证据PM2(pathogenic moderate )和PM3。结合患儿极重度聋的临床表现,推断MYO15A基因上罕见变异C.3756+1G>A和C.4519C>T (p.R1507*)构成的复合杂合突变为该患者致聋原因,故该患儿可确诊为常染色体隐性遗传性聋。
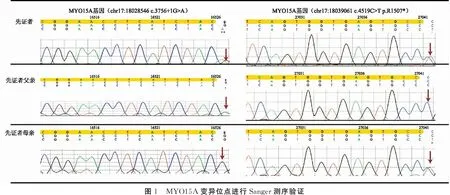
图1 MYO15A变异位点进行Sanger测序验证
3 讨论
MYO15A基因突变被认为是常染色体隐性非综合征型聋(ARNSHL)最常见的遗传原因之一,至今已有700多个基因突变被报道,并已证实有200多个变异与疾病有关,并且变异涉及多种突变类型,其中包括无义突变、剪接、缺失、插入等(http://www.hgmd.cf.ac.uk/)。
MYO15A基因包含66个外显子[8],该基因导致耳聋的临床表现为学语前非进行性重度-极重度全频听力损失,无前庭症状。MYO15A 基因编码的肌球蛋白XVa是一个含有3 530个氨基酸的大蛋白,是肌球蛋白(myosin)超家族中的一员。肌球蛋白超家族能够与细胞骨架机动蛋白丝结合,通过水解ATP生成能量从而产生运动。肌球蛋白XVa存在于人耳中,位于耳蜗及前庭毛细胞静纤毛的顶端,负责机械-电信号的转换[9],能够维持耳蜗毛细胞内肌球蛋白组织结构及毛细胞静纤毛的伸长,对维持人耳的正常听觉具有重要意义。
本研究对该家系进行了高精度临床外显PLUS的检测和分析,检测到MYO15A基因的两个杂合变异C.3756+1G>A和C.4519C>T(p.R1507*),测序结果显示这个两个变异分别遗传自患者的父亲和母亲(均为杂合状态),从而证实了该家庭中存在常染色体隐性遗传性聋基因突变,考虑该家系生育耳聋后代的概率为25%。本研究中发现的C.4519C>T(p.R1507*)位点是MYO15A基因的新致病位点,目前仅2020年伊朗人群中有一例报道[10],在我国尚属首次报道。本结果丰富了我国遗传性聋基因突变谱,对遗传性聋的诊断提供了新靶点。
该患者为双耳极重度感音性聋,为避免出现因聋致哑的情况,已进行了双侧人工耳蜗植入术,术中监测显示双耳神经反应遥测技术(NRT)反应良好,术后开机调试患儿适应性较好,正常进行听力及言语康复训练。目前认为尽早进行人工耳蜗植入是此类耳聋患者最有效的治疗方式,尤其是对于小于3岁的学龄前重度及极重度聋儿童,人工耳蜗植入可有效地促进其听力及言语能力的康复,帮助他们重返主流社会。此外,耳聋基因检测技术的发展及普及,可有效避免耳聋家庭再次生育听障儿童,对优生优育工作有着不可替代的指导作用。
