蒽醌类光敏剂的合成及光活性构效关系计算
2022-09-19童浩文高磊王立升刘旭
童浩文, 高磊, 王立升, 刘旭*
(1.广西大学 化学化工学院, 广西 南宁 530004;2.广西大学 医学院, 广西 南宁 530004)
0 引言
光动力疗法(photodynamic therapy, PDT)是最有发展前景的肿瘤治疗手段之一,具有微创性和低暗毒性等特点,可用于非肿瘤性疾病以及各种类型癌症的治疗[1]。光动力学的核心成分包括光敏剂、氧气以及特定波长的光。在有氧条件下,光敏剂暴露于特定波长的光时,光的部分能量随光子吸收后的热衰变或荧光辐射而释放出来,剩余的能量也可以转化为磷光或活性氧类,活性氧已被证明具有导致细胞凋亡和坏死的能力[2-4]。目前为止,虽然光动力疗法取得了部分成果;但调控光敏剂产生单线态氧的能力仍是待解决的关键问题之一[5],因此,开发能够被特定癌细胞环境激活且同时规避对正常细胞的毒副作用的光敏剂(激活条件如酸性环境或特定的酶)十分有必要[6-9]。
大黄酚是一种来源于中草药大黄的天然蒽醌类色素,广泛分布于我国各地[10]。这些色素除了具有多种生物活性和价格低廉的特点外,还具有极敏感的光敏性,被称为光敏剂[11];但因为其具有毒副作用强、选择性差等性质,所以不能有效区分正常组织和癌变部位,靶向能力差,限制了其在临床实践上的应用[12]。
为了解决上述问题,本文设计一系列新型可激活光敏剂(activated-photosensiter, APs),其中化合物A3在一系列表征与分析中性能较为突出,为蒽醌类光敏剂的研究提供了参考,具备一定为光动力法治疗癌症的潜在价值。
1 材料和方法
1.1 仪器和试剂
真空干燥箱(DZF-6020型,上海目尼实验设备有限公司);集热式恒温磁力搅拌加热器(DF-101SA型,上海玛尼仪器设备有限公司);旋转蒸发仪(R-1001N型,郑州长城科工贸有限公司);手提紫外分析仪(ZF-5型,上海嘉鹏仪器有限公司);磁力搅拌器(IKAMAGMS4型,上海玛尼仪器设备有限公司);电子天平(JA11002B型,上海精密科学仪器有限公司);托盘天平(JA51001B型,上海精密科学仪器有限公司);真空泵(2FY-4C-N型,郑州科工贸有限公司);双光束紫外可见分光光度计(UV-2100型,北京瑞利分析仪器有限公司);荧光分光光度计(96PRO型,上海奥析科学仪器有限公司);雷磁精密酸度计(PHSJ-4A型,上海仪电科学仪器股份有限公司);氙灯(CEL-TCX250型,北京中教金源科技有限公司);超声波清洗机(PS-100A型,上海科导超声仪器有限公司);超高效液相质谱联用仪(UPLCI-CLASS-XEVOVG2-XSQTOF型,美国WATERS公司);核磁共振仪(Bruker600MH型,德国Bruker公司)。
1.2 实验材料
大黄酚(CP, 上海源叶生物科技有限公司);对甲苯磺酰氯(AR, 上海麦克林生化科技有限公司);无水丙酮(AR, 广东光华科技股份有限公司);碳酸钾(AR, 天津大茂化学试剂厂);N,N-二乙基乙二胺(AR, 上海麦克林生化科技有限公司);乙二醇甲醚(AR, 广东光华科技股份有限公司);N-(2-氨基乙基)吗啉(AR, 上海麦克林生化科技有限公司);4-吗啉甲基苯胺(AR, 上海麦克林生化科技有限公司);3-二甲胺基丙胺(AR, 上海麦克林生化科技有限公司);三乙胺(AR, 广东光华科技股份有限公司);三氯甲烷(AR, 成都市科龙化工试剂厂);N, N-二甲基甲酰胺(AR, 上海阿拉丁生化科技股份有限公司);正己烷(AR, 广东光华科技股份有限公司);氘代氯仿(AR, 萨恩化学技术(上海)有限公司);二氯甲烷(AR, 广东汕头市西陇化工厂);乙酸乙酯(AR, 广东汕头市西陇化工厂);石油醚(AR, 广东汕头市西陇化工厂);甲醇(AR, 广东汕头市西陇化工厂);乙醇(AR, 广东汕头市西陇化工厂);二氢卟吩e6(AR, 上海源叶生物科技有限公司);ABDA单线态氧指示剂(AR, 上海源叶生物科技有限公司)。
1.3 实验方法
1.3.1 建模
定量构效关系(quantitative structure-activity relationship, QSAR)是指通过数学方法建立化合物分子描述符与其生物活性、毒性之间的线性或非线性关系模型,在分子水平上阐明化合物的分子结构与生物理化性质之间的关系[13],是一种新型药物模型,为设计结构、活性检测和毒性预测奠定基础。与传统的体外实验、体内动物实验以及传统药物活性筛选所需的有限的人体观察相比,QSAR可以大大缩短实验时间、节省人力、降低实验成本[14]。SYBYL软件可用于先导化合物的结构修饰及改造,在化合物的三维结构信息缺失的情况下,也能建立高预测能力的QSAR模型。
本文蒽醌光敏剂合成路线[15-16]如图1所示。

图1 蒽醌光敏剂合成路线
首先通过查找文献,找出具有蒽醌结构式骨架的化合物,并查找得到相应的蒽醌类化合物的半数最大效应浓度(EC50)数值[17],之后计算出它们的-ln(EC50)数值,以此数值作为化合物的毒理学指标,如表1所示。在文献中找到的化合物,首先利用Chemdraw软件画出其结构式,并将文件改成.mol格式,之后将文件导入SYBYL软件中建立数据库,同时输入-ln(EC50)数据,以活性最高的化合物作为模板分子,采用Align Database的方法对数据库中化合物分子进行叠合,叠合完毕后,对数据库表格进行刷新。添加CoMFA列,通过抽一法(leave-one-out)对数据库进行交叉验证得到交叉验证系数Q2等于0.545,最佳主成分数为5,说明模型的预测能力较好。再进行回归分析得到决定系数R2为0.908,说明模型的拟合能力较好。所建立的模型的Q2大于0.5,R2接近于1,说明所建模型具有良好的可信度。
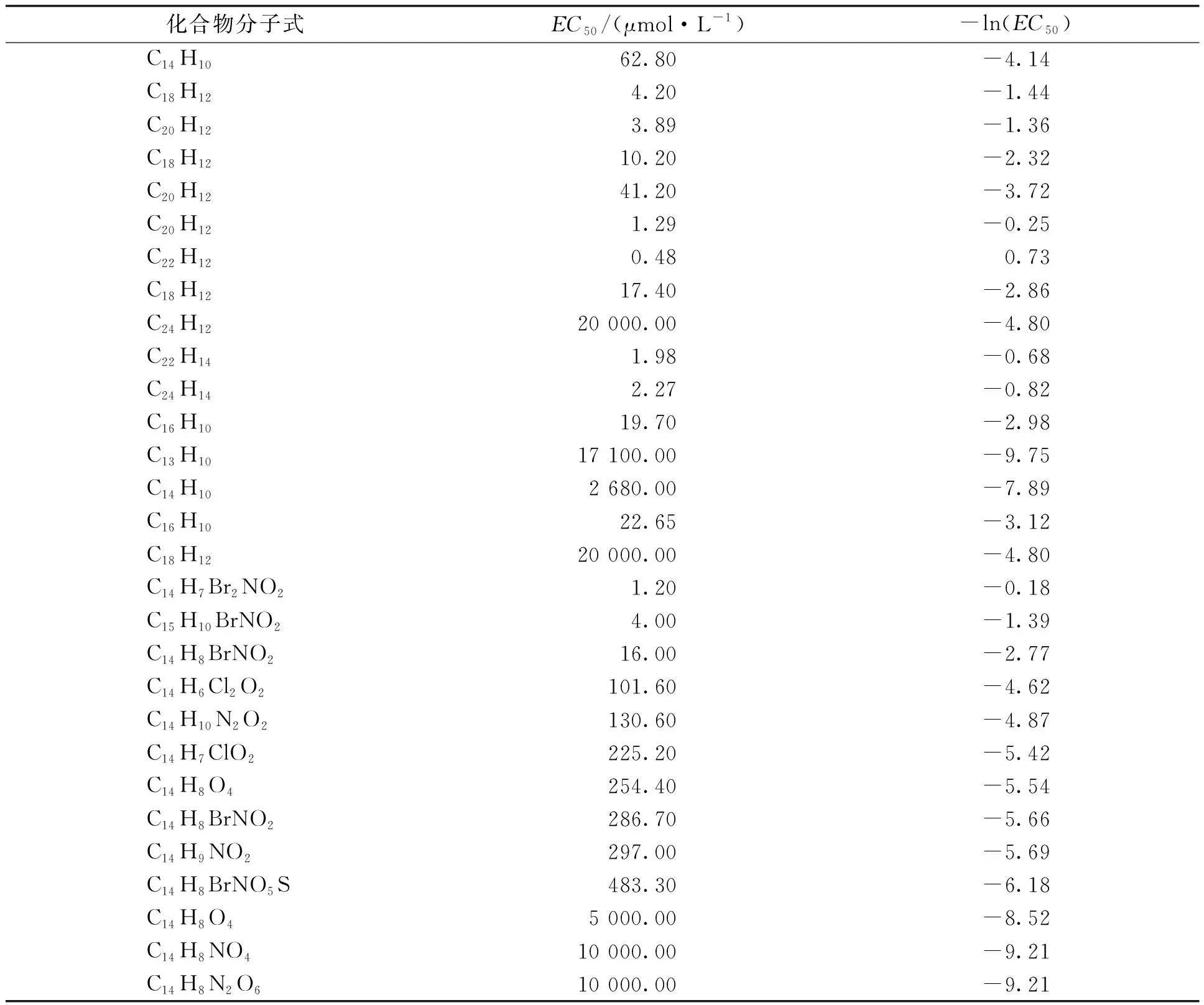
表1 蒽醌类化合物的光活性数据
1.3.2 产物A1、A2的合成
称取大黄酚(0.472 4 g,1.858 1 mmol)、碳酸钾(1.283 9 g)、对甲苯磺酰氯(1.774 1 g,9.290 5 mmol)于双颈烧瓶中(100 mL),氮气保护下,加入4 mL无水丙酮,加热至回流,反应18 h后,进行冷却。对初步产物进行旋干,加入二氯甲烷(50 mL),过滤除去固体物质杂质,再利用柱色谱法进行分离提纯[流动相:V(DCM)∶V(PE)=1∶7],得到黄色的固体产物A(0.896 7 g,1.594 mmol,收率86%)。称取产物A(200 mg,0.355 5 mmol)于双颈烧瓶中(100 mL),氮气保护下,加入乙二醇甲醚(8 mL),升温搅拌至回流,加入N-(2-氨基乙基)吗啉(0.24 mL,1.777 0 mmol),反应4 h后,冷却,旋干,经硅胶柱分离提纯[流动相:V(DCM)∶V(MeOH)=100∶1],得到红色固体产物A1(35.17 mg,0.096 0 mmol, 收率27%)、深红色固体产物A2(56.08 mg, 0.117 3 mmol, 收率33%)。
1.3.3 产物A3、A4、A5的合成
称取产物A(215 mg, 0.382 9 mmol)加入双颈烧瓶中(50 mL),氮气保护下,加入乙二醇甲醚(8 mL),升温搅拌至回流,加入N, N-二乙基乙二胺(0.28 mL),反应5 h后,冷却,旋干,经硅胶柱分离提纯[流动相:V(DCM)∶V(MeOH)=50∶1],得到紫色固体产物A3(77.64 mg,0.172 3 mmol,收率45%)、红色固体产物A4(44.62 mg,0.088 0 mmol, 收率23%)和红色固体产物A5(28.33 mg,0.080 4 mmol,收率21%)。
蒽醌衍生物A1,A2,…,A5的结构式和分子式见表2。
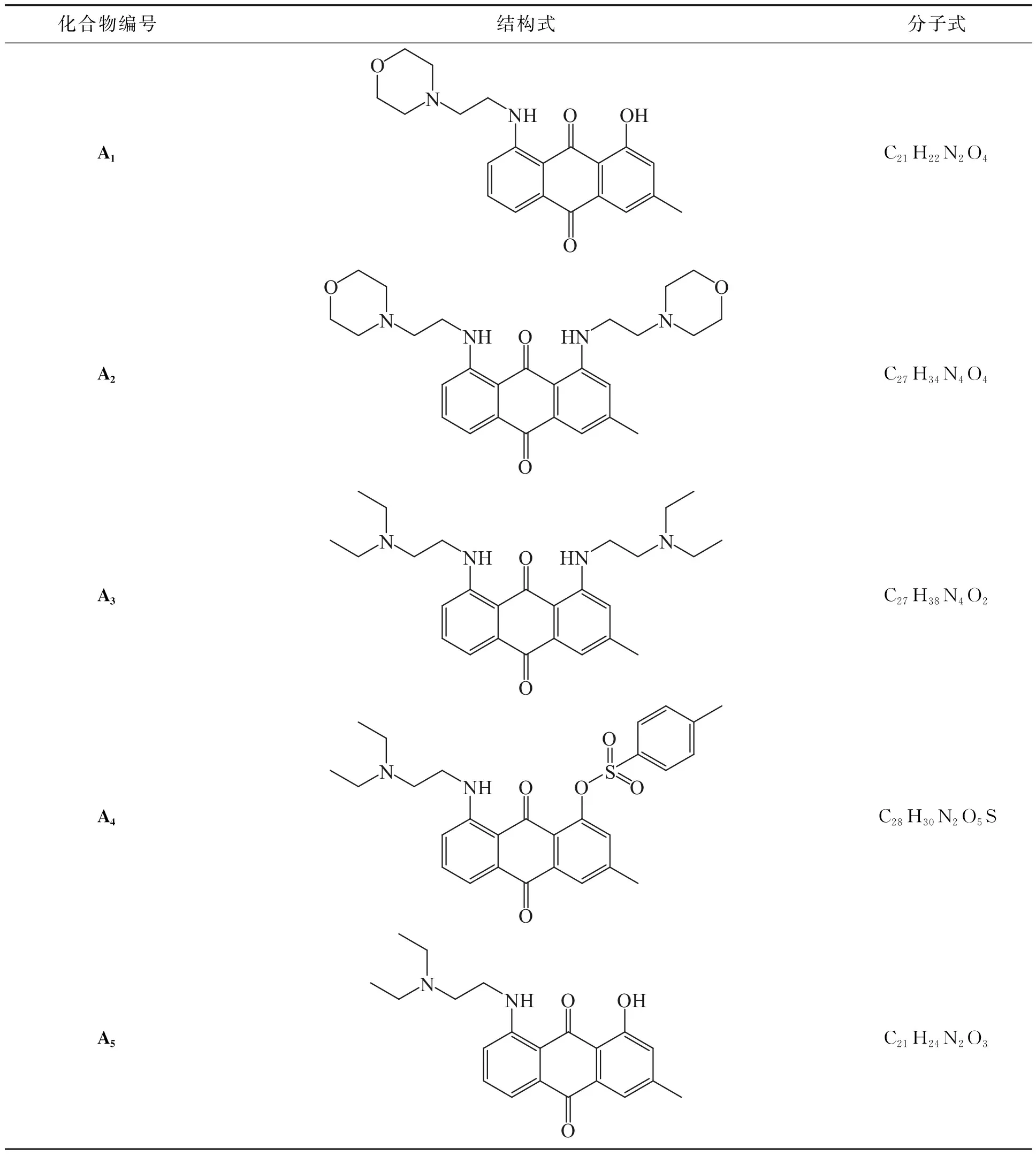
表2 蒽醌衍生物的结构式和分子式
1.4 目标化合物的理化性质及谱图数据
A1: 1-羟基-3-甲基-8-(2-吗啉乙基)氨基)蒽-9,10-二酮,红色固体,收率27%,熔点(mp)为112~114 ℃。核磁共振氢谱:1H-NMR(500 MHz, CDCl3,m=多重峰,t=三重峰,d=双重峰,s=单重峰),δ:13.16(s, 1H), 9.64(t,J=4.7 Hz, 1H), 7.75(dd,J=7.6, 1.2 Hz, 1H), 7.60~7.53(m, 1H), 7.43(d,J=1.5 Hz, 1H), 7.24(dd,J=8.3, 1.2 Hz, 1H), 6.83(s, 1H), 3.79(t,J=4.6 Hz, 4H), 3.44(q,J=5.9 Hz, 2H), 2.76(t,J=6.3 Hz, 2H), 2.58(q,J=4.8, 3.7 Hz, 4H), 2.42(s, 3H)。核磁共振碳谱:13C-NMR(126 MHz, CDCl3,s=单重峰),δ:189.65(s), 183.46(s), 162.04(s), 151.89(s), 147.17(s), 135.01(s), 134.46(s), 133.31(s), 124.19(s), 118.76(s), 117.96(s), 117.89(s), 117.30(s), 110.74(s), 67.07(s), 56.56(s), 53.40(s), 39.85(s), 29.71(s), 22.49(s), 14.13(s)。HRMS(ESI):理论计算值:([C21H22N4O2+H]+)m/z =367.165 23,实测值:m/z = 367.162 66。
A2: 3-甲基-1,8-二(2-吗啉乙基)氨基)蒽-9,10-二酮,深红色固体,收率33%, 熔点(mp)为132~133 ℃。核磁共振氢谱:1H-NMR(500 MHz, CDCl3,m=多重峰,t=三重峰,d=双重峰,s=单重峰),δ:9.76(d,J=20.6 Hz, 1H), 7.55(dd,J=7.4, 1.1 Hz, 1H), 7.48(t,J=7.9 Hz, 1H), 7.39(d,J=1.4 Hz, 1H), 7.01(dd,J=8.6, 1.1 Hz, 1H), 6.82(s, 1H), 3.80(t,J=4.6 Hz, 8H), 3.45(td,J=6.5, 4.7 Hz, 4H), 2.78(td,J=6.6, 1.9 Hz, 4H), 2.61(dt,J=7.5, 2.5 Hz, 8H), 2.41(s, 3H)。核磁共振碳谱:13C-NMR(126 MHz, CDCl3,s=单重峰),δ:188.52(s), 184.88(s), 151.15(s), 150.80(s), 145.07(s), 134.41(s), 134.17(s), 133.97(s), 117.61(s), 117.58(s), 116.46(s), 114.97(s), 114.78(s), 112.75(s), 67.00(s), 57.07(s), 57.05(s), 53.56(s), 40.07(s), 40.06(s), 29.70(s), 22.25(s)。HRMS(ESI):理论计算值:([C27H34N4O4+H]+)m/z =479.265 28,实测值:m/z =479.262 05。
A3: 1,8-二(2-(二乙氨基)乙基)氨基)-3-甲基蒽-9,10-二酮,紫色固体,收率45%,熔点(mp)为135~136 ℃。核磁共振氢谱:1H-NMR(500 MHz, CDCl3, m=多重峰,t=三重峰,d=双重峰,s=单重峰),δ:9.67(dt,J=21.9, 5.1 Hz, 2H),7.52(dd,J=7.4, 1.2 Hz, 1H),7.52(dd,J=7.4, 1.2 Hz, 1H),7.48~7.43(m, 1H),7.36(d,J=1.5 Hz, 1H),7.02(dd,J=8.5, 1.2 Hz, 1H),6.87~6.80(m, 1H),3.49(q,J=6.3 Hz, 4H),3.08~2.62(m, 12H),2.38(s, 3H),1.17(td,J=7.2, 3.8 Hz, 12H)。核磁共振碳谱:13C-NMR(126 MHz, CDCl3, s=单重峰),δ:188.38(s), 185.06(s), 151.27(s), 150.94(s), 144.85(s), 134.41(s), 134.17(s), 133.82(s), 117.64(s), 117.62(s), 116.20(s), 114.71(s), 114.67(s), 112.66(s), 51.71(s), 51.67(s), 47.24(s), 41.39(s), 41.38(s), 22.23(s), 11.90(s).HRMS(ESI):理论计算值:([C27H38N4O2+H]+)m/z=451.306 75,实测值:m/z =451.303 92。
A4: 8-(2-二乙氨基乙基)氨基-3-甲基-9,10-二氧-9,10-二氢蒽-1-基-4-甲基苯磺酸盐,红色固体,收率23%,熔点(mp)为117~118 ℃。核磁共振氢谱:1H-NMR(500 MHz, CDCl3, m=多重峰,t=三重峰,d=双重峰,s=单重峰),δ:9.57(t,J=5.4 Hz, 1H),8.01(s, 1H),7.79(d,J=8.8 Hz, 2H),7.48(d,J=5.3 Hz, 2H),7.31-7.18(m, 3H),7.10~6.97(m, 1H),3.39(q,J= 6.4 Hz, 2H),2.81(t,J=6.9 Hz, 2H),2.67(q,J=7.2 Hz, 4H),2.40(d,J=40.8 Hz, 6H),1.13(t,J=7.1 Hz, 6H)。核磁共振碳谱:13C NMR(126 MHz, CDCl3, s=单重峰),δ:183.29(s), 183.01(s), 151.17(s), 147.72(s), 145.27(s), 144.63(s), 134.74(s), 134.73(s), 133.68(s), 132.74(s), 130.58(s), 129.48(s), 129.40(s), 128.87(s), 128.83(s), 126.56(s), 125.32(s), 118.18(s), 114.94(s), 113.65(s), 51.64(s), 47.29(s), 41.40(s), 29.70(s), 21.67(s), 21.57(s), 11.87(s)。HRMS(ESI):理论计算值:([C28H30N2O5S+H]+)m/z =507.194 82,实测值:m/z =507.191 41。
A5: 8-((2-(二乙氨基)乙基)氨基)-1-羟基-3-甲基蒽-9,10-二酮,红色固体,收率21%,熔点(mp)为117~118 ℃。核磁共振氢谱:1H-NMR(500 MHz, CDCl3, m=多重峰,t=三重峰,d=双重峰,s=单重峰),δ:3.41(q,J=5.9 Hz, 2H),2.81(t,J=6.4 Hz, 2H),2.65(q,J=7.1 Hz, 4H),2.41(s, 3H),1.11(t,J=7.1 Hz, 6H)。核磁共振碳谱:13C NMR(126 MHz, CDCl3, s=单重峰),δ:189.50(s), 183.54(s), 162.01(s), 152.03(s), 147.07(s), 134.90(s), 134.44(s), 133.34(s), 124.12(s), 118.69(s), 118.00(s), 117.86(s), 117.35(s), 110.60(s), 51.30(s), 47.08(s), 29.71(s), 22.48(s), 14.13(s), 11.89(s)。HRMS(ESI):理论计算值:([C21H24N2O3+H]+)m/z =353.185 97,实测值:m/z =353.185 97。
1.5 紫外-可见光光谱
制备化合物浓度为10 μmol/L溶液[V(H2O)∶V(MeOH)=1∶1, 50 mL]。借助浓度为 5 mol/L的盐酸和5 mol/L的氢氧化钠溶液,将样品溶液pH由酸性调为碱性,即pH值分别为2.0~8.0的7组待测溶液,加入酸或碱的量应尽可能少,以避免稀释。记录波长范围为400~800 nm的波长吸收曲线,获得最大吸收的波长数据和吸光度。所有实验均在室温下使用石英比色皿进行。
1.6 荧光光谱
制备样品浓度为10 μmol/L溶液[V(H2O)∶V(MeOH)=1∶1, 50 mL]。利用浓度为 5 mol/L 的盐酸和5 mol/L的氢氧化钠溶液,将样品溶液pH由酸性调为碱性,pH分别为2.0~8.0的7组待测溶液,加入酸或碱的量应尽可能少,以避免稀释。利用荧光光度计对化合物的激发波长进行测定,确定激发波长后,从而测量不同pH值下化合物的荧光发射波长强度,所有实验均在室温下使用石英比色皿进行。
1.7 单线态氧检测
以9,10-蒽二基-二(亚甲基)二丙二酸(ABDA)作为单线态氧指示剂,以光敏剂二氢卟吩e6(Ce6)作为标准对照组。将1 mg ABDA溶于1 mL甲醇中配制质量浓度为1 g/L的ABDA指示剂溶液,避光冷藏备用。用移液枪量取2份123 μL的ABDA指示剂溶液(质量浓度为1 g/L),分别加入到2份样品浓度为10 μmol/L 的4 mL溶液中[V(MeOH)∶V(H2O)=1∶1],pH分别为5.5和7.4。以氙灯为光源,功率为250 W,对样品进行照射,每隔1 min,取样测一次紫外吸收波长,收集波长范围为300~650 nm的紫外吸光度。
2 结果与讨论
2.1 模型分析
不同角度下观察的模型视图如图2所示,图中黄色部分(虚线)表示亲水性基团,蓝色部分(实线)表示疏水性基团,减小黄色部分基团或增加蓝色部分基团能增强化合物的活性。实验中,我们对蒽醌母核上1号、8号碳位置进行了改造,分别引入疏水基团,所以,通过模型分析可以初步判断所合成的化合物具有一定的光活性。
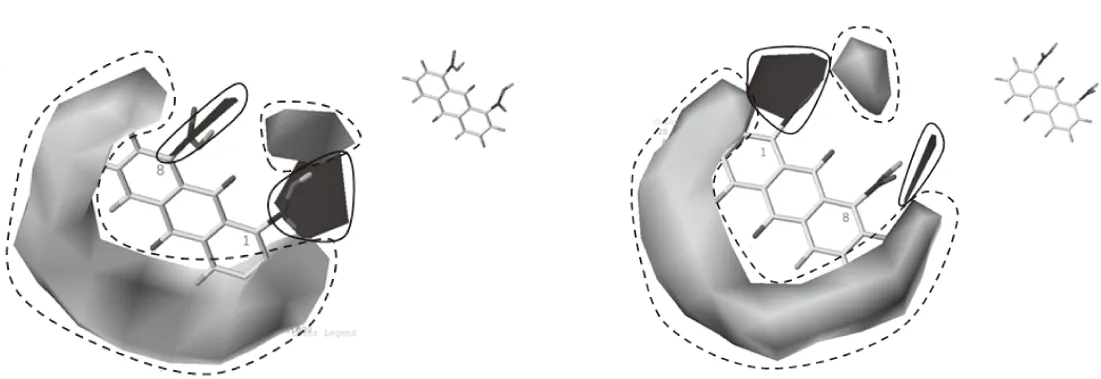
图2 不同角度下观察的模型视图
2.2 合成产物的讨论
最终化合物的合成分两步反应,首先通过酯化反应得到中间产物A,然后对产物A进行修饰,得到最终产物。
该反应具有条件比较温和、稳定、效率高且适用范围广的特点,对所合成出来的化合物经核磁共振氢谱、碳谱、质谱进行表征,产物结构得到了确证。
最终产物性质稳定,但在分离纯化时因产物点相离较近而不易分离,在对产物进行柱色谱分离后的产物还需进行重结晶等纯化步骤。由于使用了高沸点溶剂以及溶剂石油醚造成谱图有溶剂峰,不易去除,需要对产物进行再次处理以便降低溶剂峰,因此导致终产率比较低。
2.3 光物理性质
在化合物A3的合成过程中,引入可质子化的叔胺基(N,N-二乙基乙二胺),使化合物吸收最大波长相比于大黄酚发生了显著红移(图3),有利于光穿透组织激活光敏剂,吸收最大值的增加对 PDT 的应用非常有益。由于胺基质子化,化合物A3的吸收光谱在酸性 pH 值中显示出轻微的蓝移,证实了光诱导电子转移(photoinduced electron transfer,PET)机理的存在。如图3(b)所示,以530 nm为激发波长,在653 nm处观察到最大发射波长。
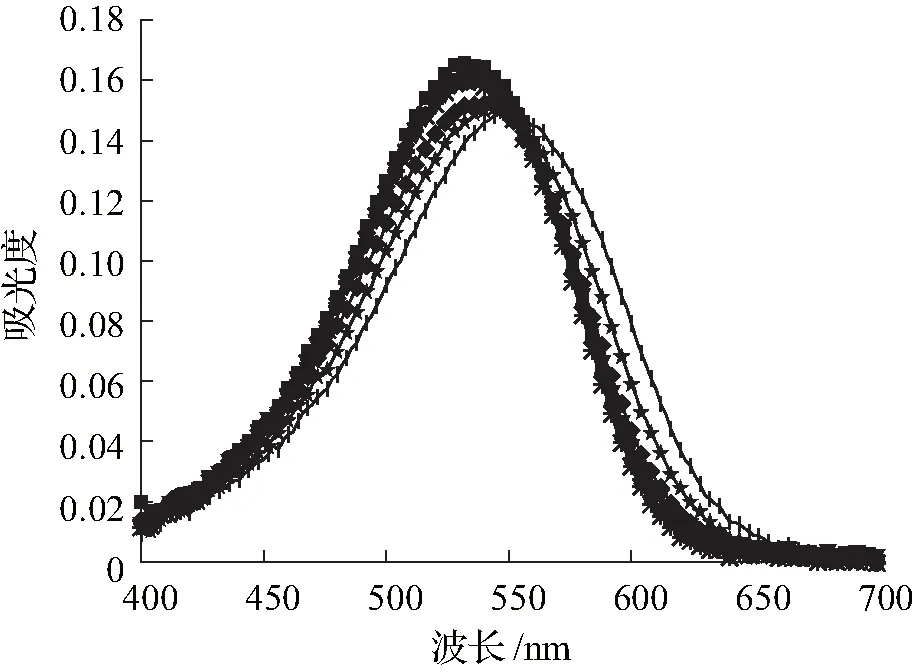
(a)紫外可见光谱
事实上,当 pH 从 8.4 变化到 2.3 时,荧光发射强度几乎增加了近 8.4 倍,进一步证实了PET机制的存在。在低pH值下观察到的化合物A3的荧光增强可归因于叔胺基的质子化,约束了 PET 过程,然后将荧光“打开”。在没有质子化的情况下,N原子的孤对电子可以转移到激发的化合物A3上,然后荧光被猝灭。对所有测量的光谱进行分析可得结论化合物A1,A2,…,A5均具有PET的性质,荧光强度随化合物的质子化程度的增加而增强。
2.4 光化学性质
目前单线态氧的检测方法包括直接检测法和间接检测法,本文所采用的检测方法为间接检测法中的紫外吸收光谱法,其原理是通过监测荧光探针与单线态氧反应后紫外吸收光谱的变化,间接检测单线态氧。本次测量实验中,以Ce6作为对照,所采用的ABDA指示剂浓度为25 μmol/L。由图4—6可知,加入ABDA指示剂测量化合物A3在不同pH环境中,随着光照时间的增长,产生的活性氧越多,紫外吸收也随之减小。而在pH=5.5的环境中,在5 min内紫外吸光度下降了84%,比在pH=7.4环境中更为显著,活性氧的产生能力明显优于对照组和其余光敏剂,且主吸收峰处的吸光度基本不发生变化,由此可见化合物A3具有较好的光稳定性。
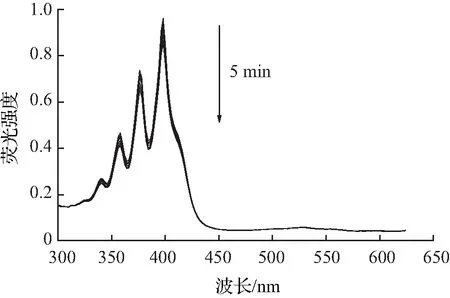
(a)pH=5.5
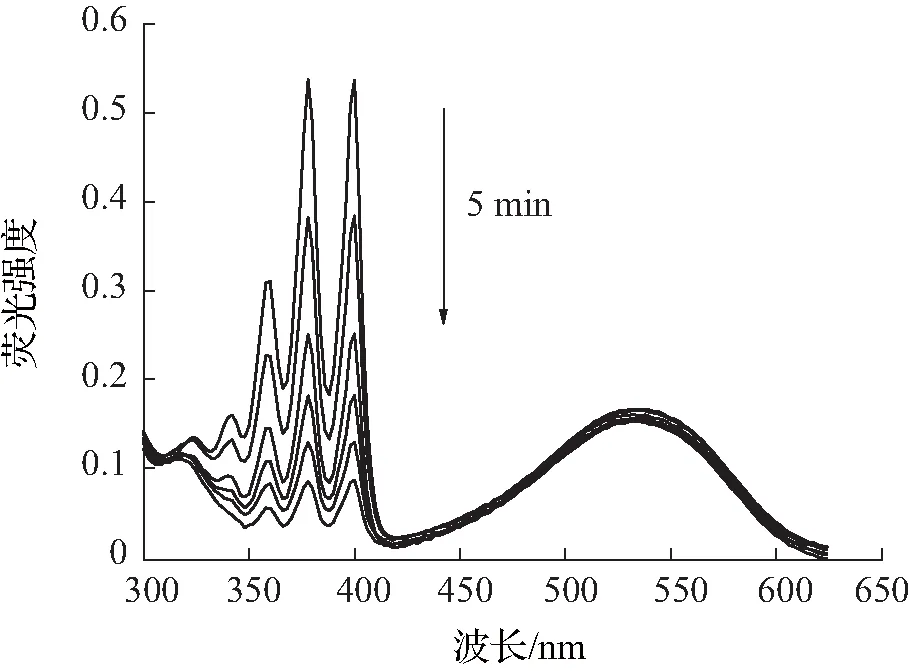
(a)pH=5.5
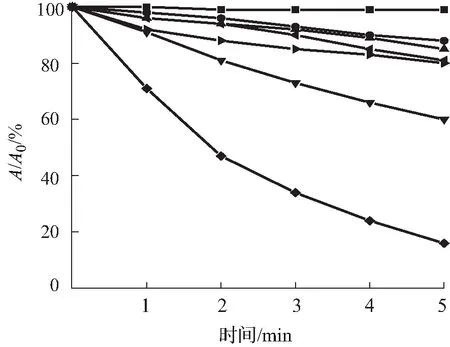
(a)pH=5.5
3 结论
本文借助SYBYL软件对数据进行处理,构建得到的蒽醌类化合物光活性QSAR模型,具有较好的拟合效果和内部预测能力,因此新型蒽醌类先导化合物的研发可基于SYBYL软件并依据关键活性位点进行设计。基于所建立的模型,设计了一系列质子活化的大黄酚衍生物。此外,通过化合物的合成、表征及光谱测定,对不同pH条件下的紫外吸收度和荧光强度进行了检测,并使用ABDA单线态氧指示剂,通过测试紫外吸收强度的变化表征单线态氧的产生,并且合成的化合物具有较强荧光性,发射峰在600~700 nm,这一范围属于红光范围,有效避开了生物自发光范围(400~500 nm)。单线态氧的检测实验中,在相同光照条件下,酸性环境中所产生的单线态氧数量比弱碱性环境中所产生的量更多,且产生单线态氧的量与模型所预测的结果一致。光动力疗法可实现精准治疗以此保证在达到治疗效果的同时减小对正常细胞组织的伤害,对蒽醌类衍生物的研究有望解决较多威胁人类健康的癌症、肿瘤问题,并且蒽醌类化合物分布广泛,材料易于获取,因此具有巨大的开发前景。
