一例产前诊断核型为45,XY;der(3:13)(p26:q10),-13 mat的健康儿童报道
2022-09-17孙丹王小艳宋婕萍
孙丹,王小艳,宋婕萍*
(1.武汉金域医学检验所有限公司,武汉 430056;2.湖北省妇幼保健院检验科,武汉 430070)
一、病例资料
患者,34岁,孕1产0,因无创DNA检测(NIPT)提示3号染色体短臂末端缺失6.6 Mb,于孕18+2周来我院就诊。患者夫妇表型正常,否认近亲结婚,否认单基因病家族史,否认辅助生殖病史。患者平素月经规律,孕期顺利,无特殊不适。孕早期胎儿颈后透明层(NT)值1.6 mm;孕24周超声提示无异常,心脏B超提示永久上腔静脉,未见其他异常;孕32周超声显示与临床孕周相符,孕期无任何其他异常。患者及其家属签署知情同意书,选择行羊水穿刺,通过核型分析和染色体微阵列技术(chromosome microarray,CMA)对胎儿染色体进行检测,并抽取患者夫妇外周血进行父母验证。染色体G显带结果显示胎儿核型为:45,XY;der(3:13)(p26:q10),-13,即胎儿有45条染色体,3号染色体和13号染色体发生易位,产生了一条衍生染色体(图1);CMA分析发现染色体3p26.3p26.1位置arr[GRCh37]3p26.3p26.1(61892-6664933)x1发生6.6 Mb的缺失(拷贝数为1)(图2)。

图1 胎儿G显带染色体核型结果 45,XY;der(3:13)(p26:q10),-13(箭头所示为异常的染色体)

图2 胎儿CMA检测结果 arr[GRCh37]3p26.3p26.1(61892_6664933)x1(3p末端显示6.6 Mb缺失,红色标记所示为缺失区域)
抽取患者夫妇外周血进行父母验证,进行核型分析和CMA检测。胎儿母亲核型为:45,XX;der(3:13)(p26:q10),-13(图3);芯片结果为:arr[GRCh37]3p26.3p26.1(61892_6634611)x1(图4)。父亲核型分析和CMA检测结果均未见异常。父母验证结果显示,该胎儿异常染色体遗传自母亲。遂对胎儿母亲进行详细评估:母亲无上述特殊面容,本科毕业,从事会计工作,无感染和手术史等。询问胎儿母亲家族史,其父母拒绝做相关检测,其弟弟和妹妹均已结婚且正常生育,推测该孕妇异常染色体可能为新发突变,但不排除遗传自携带有平衡易位的父母。经过遗传咨询,夫妇双方选择继续妊娠,于39+4周剖腹产分娩一男婴,出生时体重3 350 g,身长52 cm,Apgar评分良好,样貌等未见异常,新生儿筛查结果均未见异常。两年后对该家庭进行随访,男孩母亲表示该男孩1岁左右开始走路,目前语言表达正常,未见3p缺失综合征相关临床表现,也无特殊面容等。
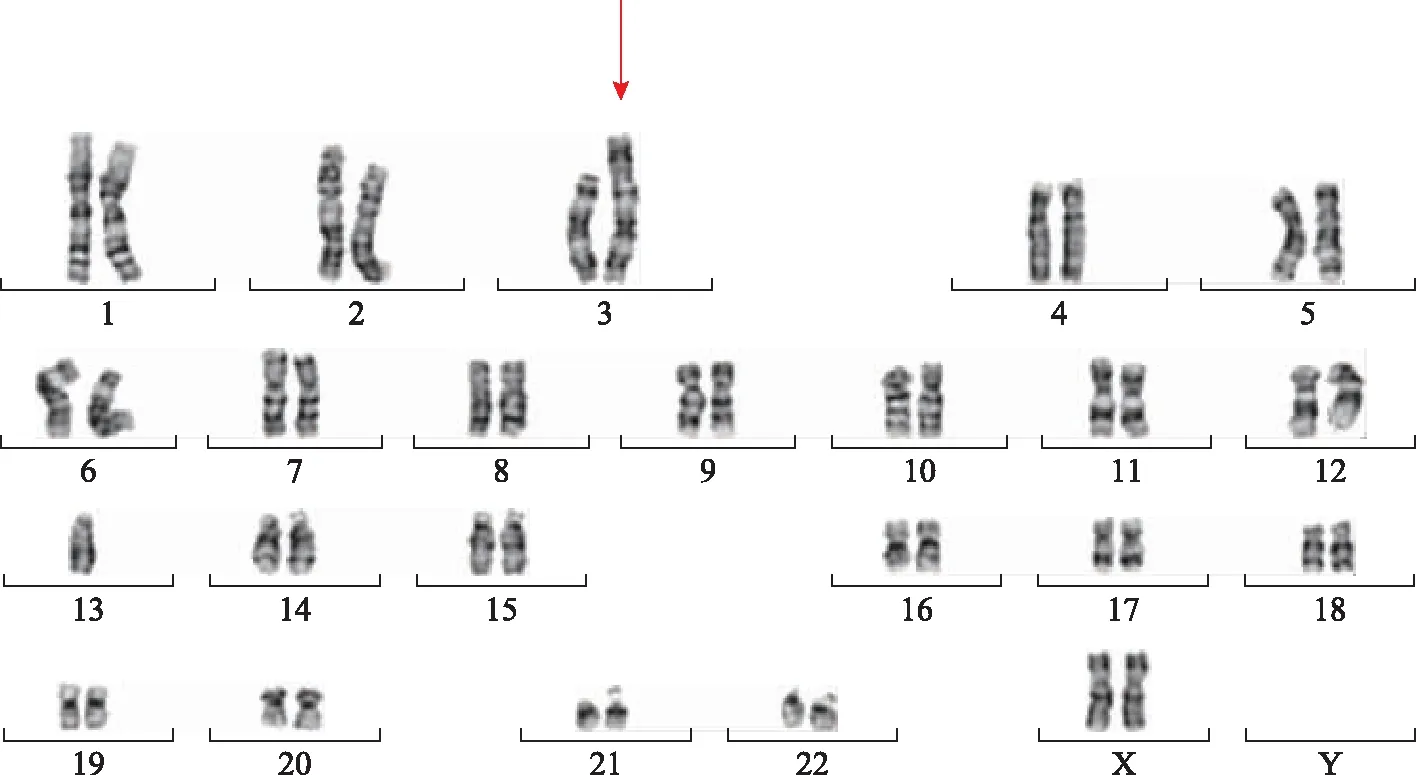
图3 母亲G显带染色体核型结果 45,XX;der(3:13)(p26:q10),-13(箭头所示为异常的染色体)
二、讨论
3p缺失综合征是一种罕见的由3p25区域到末端区域缺失引起的染色体疾病,缺失大小可从1 Mb到几Mb不等,过往的病例报道不存在共同的断裂位点,具体致病基因未知,多数为新发突变,也有少量家族性病例报道[1-4]。该综合征通常会导致智力障碍、发育迟缓和其他异常等,通常表型异质性较大。其他常见的特征还包括内眦褶皱、多指及腭裂、癫痫发作、肌张力降低、肠道异常或先天性心脏病等[5-6]。3号染色体短臂的末端仅包含少量的基因,根据以往的病例报道,推测与该综合征表型相关的基因可能有CHL1(OMIM-ID:607416)、CNTN4(OMIM-ID:607280)、CRBN(OMIM-ID:609262)、CNTN6(OMIM-ID:607220)等[7-8]。
本文中的病例通过核型分析和染色体微阵列技术(CMA)检测在产前诊断为3p末端存在6.6 Mb的片段缺失,该区域共包含41个基因,包含 CHL1、CNTN6、CNTN4、IL5RA、TRNT1、CRBN、LRRN1、SETMAR、SUMF1、ITPR1、BHLHE40、ARL8B、EDEM1等13个蛋白编码基因,在正常人数据库中无报道,该区域内无明确的单倍剂量敏感基因,decipher数据库中有超过40多例与该样本重叠的病例报道,有些是新发拷贝数变异,有些遗传自表型正常的父母,有些遗传自平衡易位的父母,但表型高度可变且不具有特异性,最常见的表型为面部畸形和语言发育迟缓等。本病例包含以往报道过的责任基因,异常核型为45,XY;der(3:13)(p26;q10),-13,且遗传自表型正常的母亲,胎儿出生后与母亲一样无特殊表型异常。
CNTN4(OMIM:607280)基因是一种重要的突触粘附蛋白,属于Contactin超家族,人类遗传学与小鼠模型证据表明,突触形成和结构缺陷与包含自闭症在内的神经发育障碍密切相关,多条证据表明CNTN4与自闭症的风险有关。已有多篇文献报道该基因的拷贝数变异与癫痫、精神分裂症、注意缺陷多动障碍、双相情感障碍、视神经再生障碍、原发性开角型青光眼、脊髓小脑共济失调有关[9-10]。编码Contactin家族另一成员的CNTN6(OMIM:607220)基因,定位于CNTN4基因的远端,参与少突胶质细胞的生成,在小脑中表达水平较高,CNTN6基因的拷贝数变异在癫痫、智力低下和发育迟缓的病例中存在较多报道[11-12]。然而,Coe等[13]对29 085名发育迟缓儿童与19 584名健康对照组进行了拷贝数变异发病率对比,结果显示,神经发育障碍患者CNTN6拷贝数变异的发生率为0.4%,正常儿童的发生率为0.3%,这些拷贝数变异可以在“健康个体”中检测到,可能因为具有轻度表型的个体无法识别,同时也不排除这些CNV是神经发育迟缓的风险因素的可能性。Contactin基因家族在整个脊椎动物进化过程中相当保守,这表明该蛋白家族对于神经发育可能是必不可少的。另一个基因CHL1(OMIM:607416),编码免疫球蛋白超家族的细胞粘附分子,在神经元中高度表达,CHL1+/-和CHl1-/-基因敲除小鼠模型提供了CHL1可能导致与“3p缺失综合征”相关的精神损害的证据。多个仅包含CHL1基因拷贝数变异的病例报道显示CHL1基因的单倍剂量不足会导致患者发生神经发育迟缓[14-16]。CRBN(OMIM:609262)基因是一个与认知相关的基因,它参与人类记忆和学习的过程,常呈常染色体隐性遗传模式,该基因的完全的失功能性会导致严重的智力低下、发育迟缓、癫痫等。由于无法检测到点突变、动态突变等其他异常类型,CMA通常无法对隐性遗传病进行确诊,该基因的拷贝数变异合并该基因等位基因的点突变也许是引起有症状的3p缺失综合征的病因[17-19]。ITPR1(OMIM:147265)基因是一种调节细胞内钙信号传导的门控钙通道,该基因的突变与脊髓小脑共济失调相关,呈常染色体显性遗传模式,且变异类型多为错义突变,推测其可能机制为显性负性效应;该基因的突变也与Gillespie综合征相关,表现为:双侧虹膜对称部分发育不全、固定的大瞳孔、小脑发育不全伴共济失调、先天性肌张力减退和不同程度的智力障碍等,存在显性或隐性遗传模式,推测其可能也具有显性负性效应,推测该基因的单倍剂量不足可能不足以引起相应病症[20]。然而,文献中报道了大量包含在上述区域内的拷贝数变异而表现出自闭症、癫痫等症状的病例报道,同时也存在遗传自表型正常父母的相关病例报道,因而增加了相关基因拷贝数变异的遗传咨询的难度。同时在神经发育和神经系统疾病中,存在不完全外显和表型高度可变的现象,常检测出类似于本病例中临床意义不明的拷贝数变异,这给产前遗传咨询造成了困扰。
在产前诊断中,CMA作为一个应用多年的一线诊断技术,与传统的细胞遗传学检测技术相比,具有全基因组覆盖、无需细胞培养、分辨率较高等优点,但同时会检测到许多临床意义不明的拷贝数变异,还会出现携带有相同的拷贝数变异的个体呈现出不同表型的现象。一些遗传自表型正常父母的拷贝数变异可能会导致后代出现一些异常表型,如存在多例出现在3p末端区域内缺失而表现出异常的病例报道,通常认为这些拷贝数变异具有外显不全等特点,这对于产前诊断是一个巨大的挑战。为了解释这种CNV相关的临床异质性,目前已经提出了3种可能的机制:(1)存在额外的CNV(“二次打击”学说);(2)该拷贝数变异包含隐性遗传病的基因;(3)该拷贝数变异是某些疾病的易感因素,存在影响表型的表观遗传或其他环境因素等等[21-23]。但由于其复杂性,上述3种可能机制较难应用在临床实践中,未来需要更多相关研究来阐明其可能机制。本案例通过产前诊断明确了一例携带有遗传自母亲的异常染色体,且出生后与母亲一样无明显异常表型的病例报道,表明该区域内包含的基因可能不是3p缺失综合征的关键基因或存在其他引起3p缺失综合征的因素等。
由于平衡易位的携带者本身可能由于基因总数未丢失仅是染色体片段出现位置改变而不表现出表型异常,但在减数分裂产生配子时可产生不平衡的配子。该病例中母亲携带的不平衡的染色体推测可能来源于其发生了平衡易位的父母,但由于孕妇父母拒绝接受核型分析检测,因而无法进行验证。通过了解孕妇家族史,孕妇家族其他成员未发生不良生育及家族史等,该孕妇易位的不平衡染色体也有可能是父母在形成生殖细胞的过程中的随机事件,具体不详。
通过本病例,我们推测3p末端缺失综合征的关键基因可能不存在于3号染色体短臂末端6.6 Mb内,但不排除外显率不全或携带者表型较轻而未被观察到的可能性,拷贝数变异作为遗传变异的一个重要来源,对其评估是一个相对复杂的过程,还存在许多未知的问题,未来还有更多需要探索的内容。希望报道更多的相关病例,以帮助医务工作者理解这类存在争议的拷贝数变异的临床意义。
