基于生物信息学技术的非梗阻性无精子症关键microRNAs和基因的研究
2022-09-17赵铭佳郭剑耿女苏兴李翠姚雨宏晏红伟韩宝生刘美玲卢文红谷翊群
赵铭佳,郭剑,耿女,苏兴,李翠,姚雨宏,晏红伟,韩宝生,刘美玲,卢文红,谷翊群*
(1.唐山市妇幼保健院生殖遗传科,唐山 063000;2.北京协和医学院研究生院,北京 100730;3.国家卫生健康委科学技术研究所 男性生殖健康重点实验室,北京 100081)
全球范围内大约8%~12%的夫妇受到不孕症困扰,其中男性因素大约占50%[1]。男性不育以无精子症最为严重,分为梗阻性无精子症(obstructive azoospermia,OA)和非梗阻性无精子症(non-obstructive azoospermia,NOA)[2]。NOA病因复杂,大约73%的NOA病因不明[3]。精子发生是一个极其复杂的细胞分化过程,涉及调节生殖细胞发育和成熟的基因就多达2 300个,其中微小RNA(microRNA,miRNA)在转录后调节基因表达[4]。在以往的研究中,虽然已经提出了许多与精子发生相关的候选基因,但许多候选基因尚未得到验证或者在人群中非常罕见。研究表明,miRNA可能通过调节靶向基因mRNA的表达介导生精细胞的发育,参与精子发生,与男性不育有关[5]。本研究采用生物信息学的方法进行数据挖掘和分析,探讨非梗阻性无精子症相关的miRNAs和关键基因。
材料和方法
一、数据库检索
在NCBI的GEO数据库(网址:https://www.ncbi.nlm.nih.gov/geo/)进行基因芯片数据检索,确定检索关键词为“非梗阻性无精子症(non obstructive azoospermia)”和“智人(Homo sapiens)”。在PubMed、Embase和Web of Science中进行检索,确定检索词为“非梗阻性无精子症(non obstructive azoospermia)”和“miRNA”或“microRNA”或“ncRNA”或“micro ribonucleic acid”。筛选并下载GSE145467和GSE108886表达谱数据集。GSE145467数据集,由Hodzic等[6]提供,基于Agilent-014850全人类基因组微阵列4x44K G4112F(特征编号版本)的GPL4133平台,包括10个NOA样本和10个OA睾丸样本。GSE108886数据集,由Baksi等提供,基于Illumina HumanHT-12 V4.0表达微芯片的GPL10558平台,包含 8个NOA样本和4个OA样本(包括1个睾丸对照样本)。
本研究中所用数据库及检索筛选流程见图1。
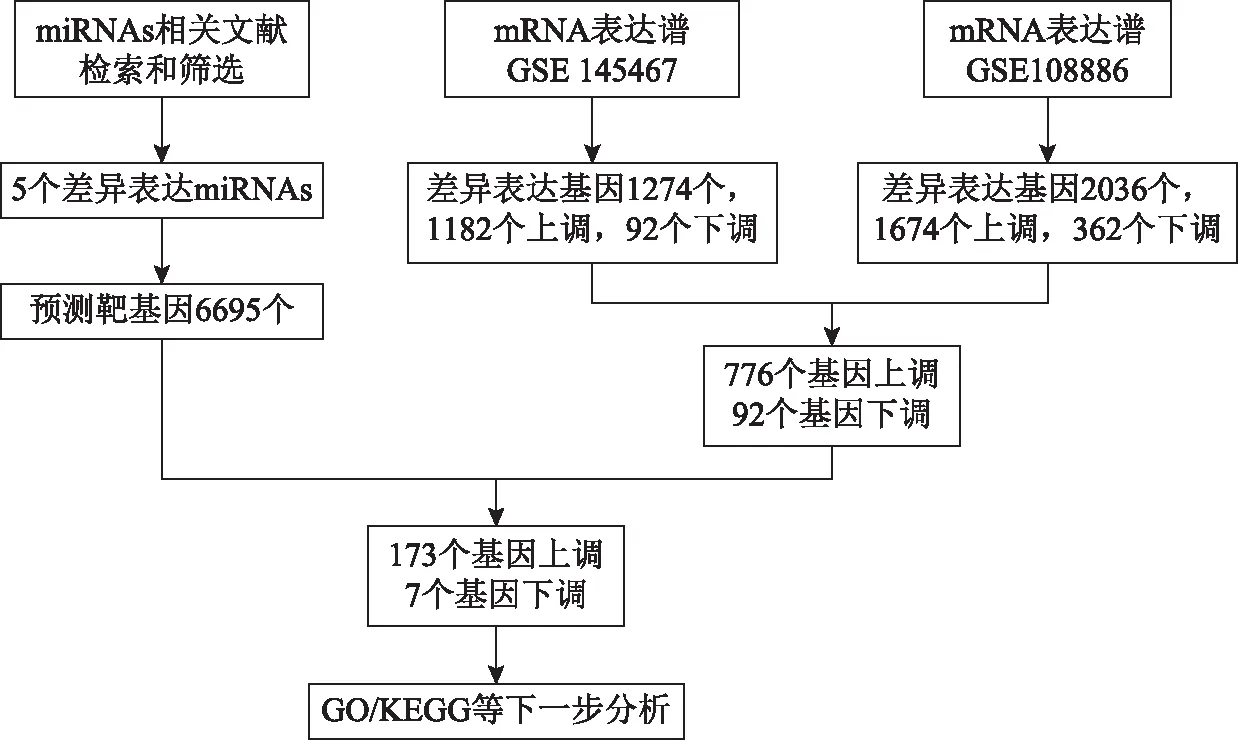
图1 基因数据库筛选及分析流程图
二、筛选差异表达miRNAs:在GEO数据库搜索已发表的NOA的miRNA数据集合,但是无可用的数据。然后,在PubMed、Embase和Web of Science中进行检索,以文献中报道非梗阻性无精子症睾丸组织中存在差异表达的miRNA并且经过PCR验证为文献筛选标准,共筛选出五项包含miRNA的研究(表1)[7-11]。该五项研究均确定了miRNA的变化趋势,再以至少两篇文献报道且经PCR验证为差异表达的miRNAs作为研究对象的miRNA筛选标准,筛选出差异表达miRNA。为研究关键miRNA在非梗阻性无精子症中的作用,选择具有一致表达变化趋势的miRNAs作为本研究的候选差异表达miRNAs用于下面的分析。
三、差异表达miRNAs的靶基因预测:将上述筛选的miRNAs汇总,通过联川生物云平台的“miRNA靶基因预测云分析”(https://www.omicstudio.cn/analysis/targetGene)预测其靶基因。
四、筛选差异表达基因:利用GEO2R工具(https://www-ncbi-nlm-nih-gov-s.webvpn.cams.cn/geo/geo2r/)在GEO数据库中GSE145467和GSE108886表达谱数据集的NOA和匹配的OA中筛选差异表达基因。GSE145467筛选标准是FDR小于0.05和log2FC的绝对值大于2;GSE108886的筛选标准是FDR小于0.05和log2FC的绝对值大于1。对上述两个数据集进行Venn Diagram分析以确定差异表达基因的交集,再应用联川生物云平台(https://www.omicstudio.cn/tool/6)与miRNAs预测靶基因取交集,获得最终鉴定的差异表达基因并绘制韦恩图。
五、差异表达基因的GO和KEGG富集分析:利用注释可视化集成发现数据库(The Database for Annotation,Visualization and Integrated Discovery DAVID)(https://david.ncifcrf.gov/)进行GO功能和KEGG通路富集分析以确定常见差异表达基因的功能。选择物种为智人(Homo sapiens)。P<0.05表示差异有统计学意义。
六、差异表达基因的蛋白质相互作用网络和枢纽基因鉴定:利用基因相互作用检索工具(the Search Tool for the Retrieval of Interacting Genes STRING)数据库(https://cn.string-db.org/)构建差异表达基因的蛋白质相互作用(protein-protein interaction PPI)网络,同时要求交互分数不低于 0.7。然后,应用 Cytoscape 软件v3.7.1(https://cytoscape.org/)使源自STRING数据库的PPI网络可视化。应用 Cytoscape 中的 cytoHubba 插件对PPI 网络中的节点根据度数计算方法进行排序,前20个基因被认为是枢纽基因。P<0.05被认为具有统计学意义的差异。
结 果
一、NOA相关的差异表达miRNAs
在PubMed、Embase和Web of Science中共筛选到5项研究中差异表达并经过验证的miRNAs共56个(表1)。经过对文献结果中差异表达的miRNAs绘制韦恩图(图1),最终筛选出5个关键的候选miRNAs,其中上调表达1条为miR-10b-5p,下调表达4条分别为miR-34b-5p,miR-34b-3p,miR-34c-5p,miR-449a(表2)。
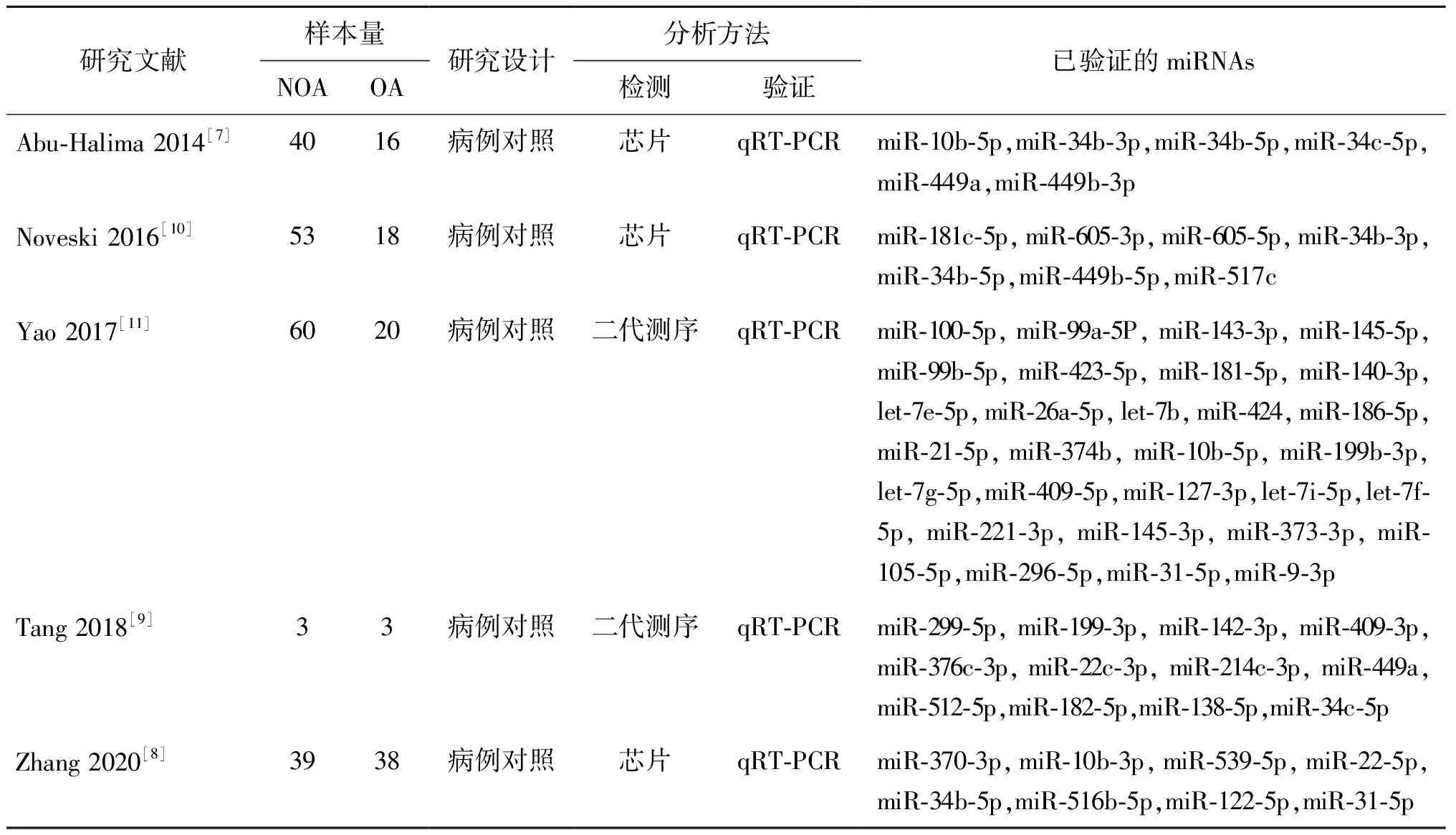
表1 筛选出的5项研究基本资料

图2 五项研究差异表达的56个miRNAs的韦恩图

表2 筛选出的5个候选miRNAs情况
二、NOA相关的差异表达基因筛选结果
通过联川生物云平台的“miRNA靶基因预测云分析”共预测靶基因6 695个。在GSE108886数据集中筛选出了2 036个差异表达基因,包括1 674个上调基因和362个下调基因。在GSE145467数据集中筛选出1 274个差异表达基因,包括1,182个上调基因和92个下调基因。对上述两个数据集进行Venn Diagram分析取交集,共鉴定出797个差异,包括776个上调基因和21个常见的下调基因(图3)。再将上述数据集的DEGs与预测获得的miRNAs靶基因取交集,最终鉴定DEGs共180个,其中上调基因173个,下调基因7个(图4)。筛选流程详见图1。
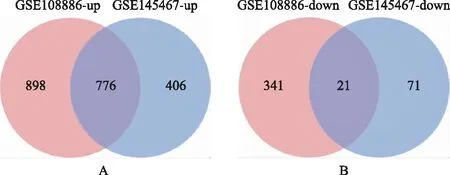
A:上调基因;B:下调基因。
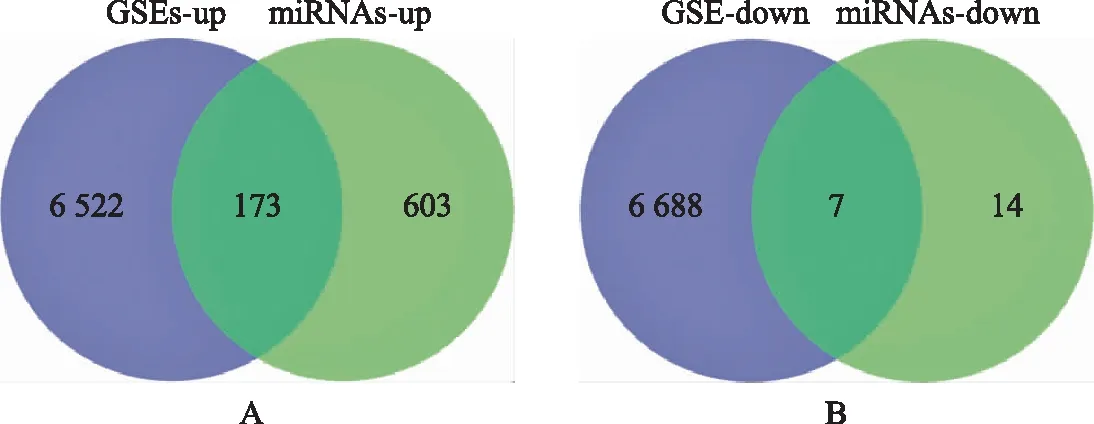
A:上调基因;B:下调基因。
三、差异表达基因的GO和KEGG分析
差异基因GO富集于生物过程(BP),主要包括纤毛组装,精子轴丝组装,鞭毛相关的精子活力,纤毛依赖性细胞运动,顶体组装,精子发生,精子细胞发育,血小板脱颗粒,细胞蛋白质代谢过程,蛋白质稳定化等;细胞组成(CC)主要包括活动纤毛,轴丝,精子鞭毛,中心粒,细胞外空间等;分子功能(MF)主要包括蛋白质结合,纤毛蛋白轻链的结合,整合素结合等(图5)。
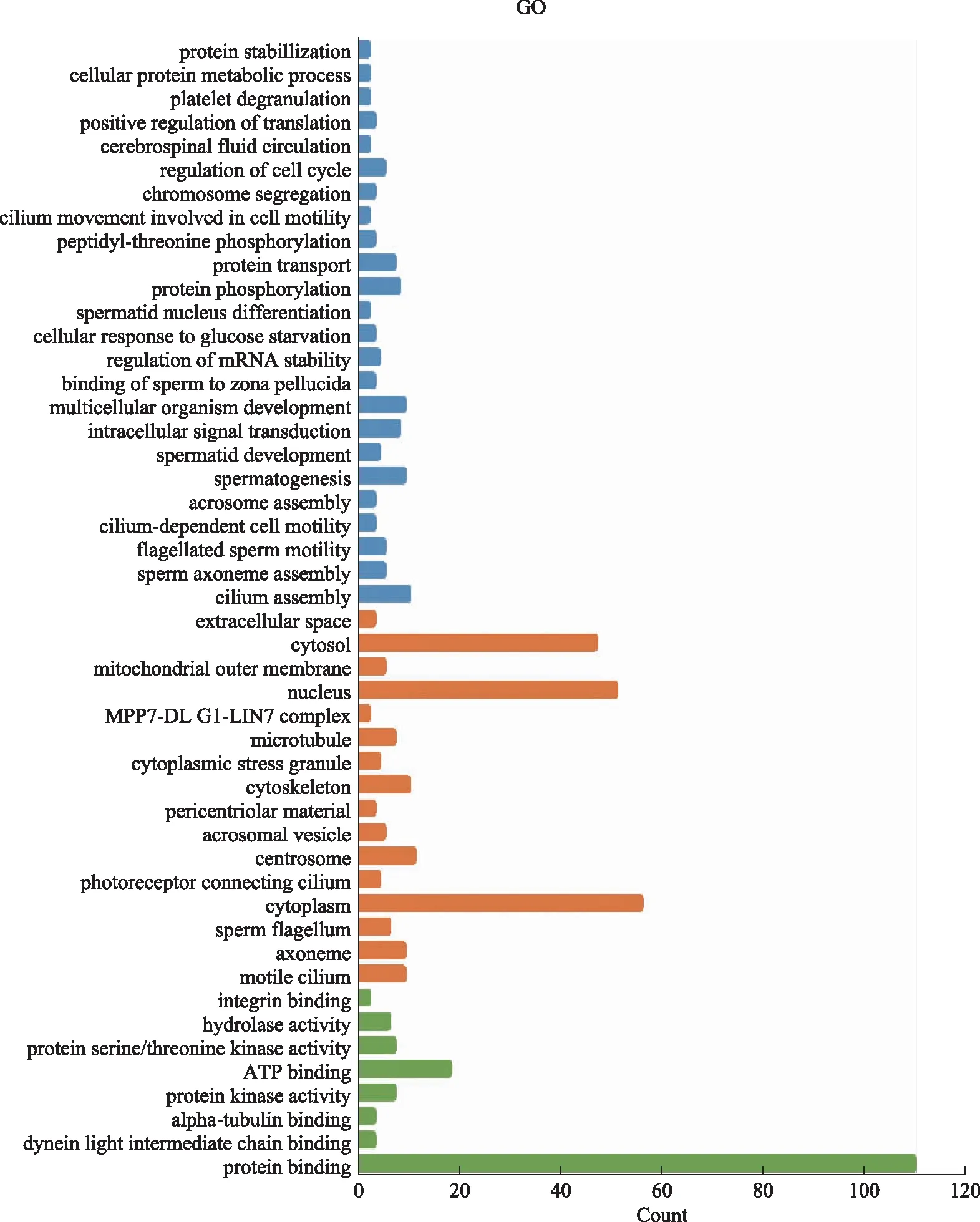
蓝色代表生物过程(BP);桔红色代表细胞组分(CC);绿色代表分子功能(MF)。
差异表达基因富集的KEGG通路主要包括卵巢类固醇激素生成,多个物种长寿调控途径,扩张型心肌病,内分泌抵抗,孕酮介导的卵母细胞成熟等(图6)。
四、差异表达基因的PPI网络和枢纽基因
通过STRING数据库构建出差异表达基因的PPI网络(图6)。Cytoscape 中的 cytoHubba 插件筛选出了排名前20的枢纽hub基因,分别为TEKT3、EFHC1、DYNLL2、DNAH2、CETN1、SPATA7、ASRGL1、CCDC146、PLCZ1、SPAG16、DNAL1、EFCAB11、SPA4L、LIN7A、TEKT1、FXR1、RPGRIP1、DPY19L2、DDX25、ZC3H14,及其相互关系(图7)。
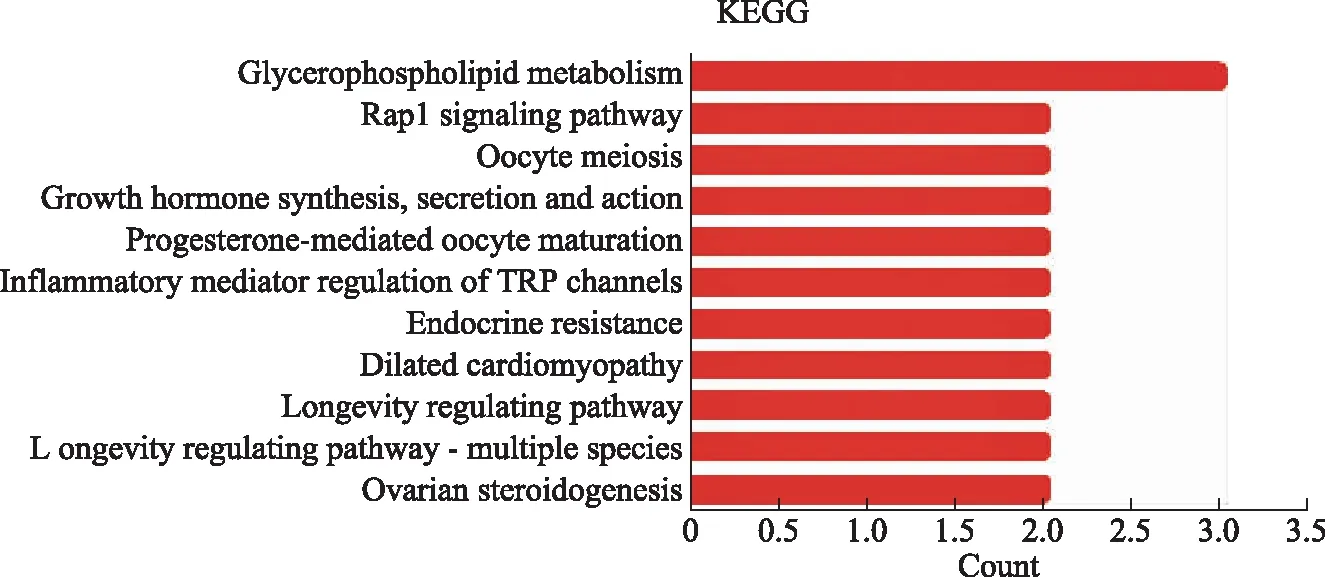
图6 差异表达基因的KEGG分析
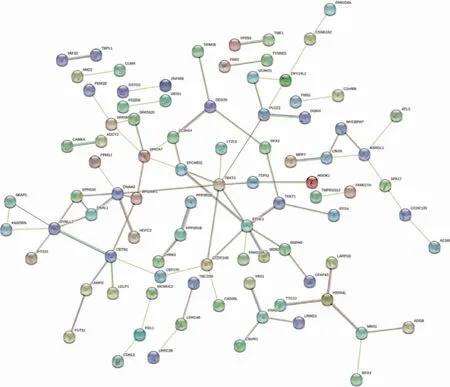
圆圈代表基因,连线代表基因之间的联系。红线:二者之间相互融合;绿线:二者之间互为接近;蓝线:二者之间同时出现;紫线:实验获得;黄线:文本挖掘获得;淡蓝色线:数据库资料获得;黑线:共表达。
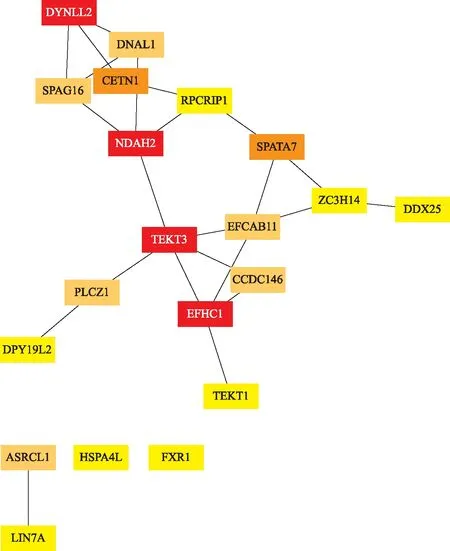
从红色到黄色,颜色越深说明相关程度越高。
讨 论
现已证明,miRNA在精子发生和发育调节中发挥作用[12]。本研究经过数据库及文献的筛选,共确定了5个NOA相关的差异表达miRNAs,其中上调表达1个为miR-10b-5p,下调表达4个分别为miR-34b-5p,miR-34b-3p,miR-34c-5p,miR-449a。
本研究显示NOA患者睾丸组织中miR-10b-5p表达下调。Wu等[13]研究发现miR-10b-5p在拮抗缺氧诱导的心肌细胞凋亡方面发挥一定的作用,可以推断miR-10b-5p可能在精子发生过程中具有一定的作用。miR-34b-5p,miR-34b-3p和miR-34c-5p均属于miR-34家族,已被证实在精子发生过程中发挥重要作用[14-15]。有研究发现miR-34b和miR-34c的靶基因dazl参与调控小鼠生殖细胞分化[16],也有研究证明miR-34c通过调控Nanos2 破坏精原干细胞的稳态[17]。巧合的是,本研究确定的miR-449a所属的家族与miR-34家族已被证明在结构上相似[12]。同时有研究表明miR-449家族和miR-34b/c 在小鼠睾丸雄性生殖细胞发育的调节中发挥着相应的作用[18]。Marcet等[19]研究证明,miR-449a的表达受E2F1的正向调节,从而促进细胞周期进程并诱导受损细胞凋亡,E2F1的过表达导致精母细胞凋亡增加。因此,本研究筛选的5个差异表达miRNAs在精子发生过程中发挥重要作用。
本研究发现差异基因GO富集于生物过程(BP),主要包括纤毛组装,精子轴丝组装,细胞蛋白质代谢过程,精子发生等方面。细胞组成(CC)主要包括活动纤毛,轴丝,精子鞭毛,中心粒等。分子功能(MF)主要包括蛋白质结合,纤毛蛋白轻链的结合等。上述结果表明差异表达基因涉及到精子发生中的很多方面,上述的某个或多个环节出现问题就有可能导致精子发生障碍,给我们后续针对5个miRNAs进行功能研究时提供了方向。在NOA相关的差异表达基因所涉及的生物学功能中,精子发生严重受损是NOA的一个特征[20]。而本研究中差异表达基因的KEGG通路主要包括卵巢类固醇激素生成、多个物种长寿调控途径、扩张型心肌病、卵母细胞减数分裂以及孕酮介导的卵母细胞成熟等,上述大多数通路参与精子的发生[21-22],且均有基因ADCY3和IGF1参与。本研究富集到的通路中“卵母细胞减数分裂”和“孕激素介导的卵母细胞成熟”为女性生殖相关的途径,与Hu等[23]进行的类似研究中得到了相同的结论。因此,“卵巢类固醇激素生成”途径的某些基因也可能在精子发生中发挥作用。
本研究中差异表达基因的PPI网络前20位的Hub基因中,Tektins(TEKTs)是一个高度保守的微管蛋白质家族,已鉴定出五种 TEKT(TEKT1、2、3、4和5)。本研究的hub基因中主要包括TEKT1和TEKT3。有报道发现TEKT3在精子中主要与线粒体表面和中间部分的外部致密纤维相关,并且TEKT3被发现存在于精子头部顶体膜的赤道段区域[24],说明TEKT3 不仅可以作为精子活力所需的鞭毛成分起作用,还可能参与顶体反应。另外,Oiki等[25]研究发现TEKT1存在于精子鞭毛和顶体帽的顶端区域,而且在小鼠精子体外顶体反应后,顶体相关的TEKT1消失。上述研究均说明了TEKT3和TEKT1在精子发生过程中的重要角色。本研究中与精子鞭毛相关的基因还包括EFHC1、DNAH2和FXR1。Li等[26]在研究中证明EFHC1在精子鞭毛组装相关蛋白中发挥重要作用。关于DNAH2基因即编码动力蛋白轴丝重链2,在最近的一项男性不育畸形精子症的全外显子组测序的研究中发现DNAH2的突变导致精子鞭毛的形态异常[27]。Huot等[28]在研究中观察到成年小鼠睾丸中发现了最高水平表达的FXR1与微管元件相关。关于基因DYNLL2,虽然目前还未见其编码动力蛋白轻链LC8型2与男性生殖系统关系的研究报道,然而,Hu等[23]在类似的研究关于hub基因的预测中也得出了DYNLL2基因。本研究中与精子纤毛相关的基因主要包括SPATA7和SPAG16。SPATA7基因编码一种纤毛蛋白,通常在光感受器和精母细胞中表达[29],SPATA7 基因的突变很可能会导致非梗阻性无精子症。有研究已经证明SPAG16基因功能的丧失会导致雄性不育和严重的精子活力缺陷,同时SPAG16基因的杂合突变降低了精子轴突中央装置的稳定性[30]。本研究中发现中心体相关的基因主要包括CETN1和CCDC146。Avasthi等[31]在关于雄性小鼠的中心体蛋白(centrin)的研究中证实CETN1在精子发生和精子细胞成熟的后期步骤中发挥重要作用。Firat-Karalar等[32]在通过哺乳动物精子细胞的中体粒蛋白质组质谱鉴定的研究中发现CCDC146基因编码中心体相关蛋白。PLCZ1已被证明是一种关键的精子卵母细胞激活因子的编码基因,PLCZ1基因的突变已被证明会导致男性不育[33-34]。DPY19L2是导致精子头部畸形的主要基因之一[35],Castaneda等[36]研究发现DPY19L2是小鼠精子顶体发生和生育力所必需的基因。DDX25是促性腺激素和雄激素作用的靶点,是精子发生基因的转录后调节关键因子[37]。通过对上述hub基因的分析讨论充分说明了这些基因在精子发生过程中的重要意义,同时为下一步的研究提供了新的方向。
本研究存在一些局限性:(1)对miRNAs的筛选的文献质量有区别,样本量和实验方法也存在一定的区别。(2)对于筛选差异表达基因的数据均是来源于GEO数据库的芯片数据,样本量较小,样本的质控等方面均对结果存在影响。(3)关键的miRNAs和hub基因需要通过实验来进一步的研究验证。(4)精子发生是一个动态过程,该分析仅在一个时间点提供有关基因表达和精子发生状态的信息。
本研究通过一系列的生物信息学分析,结果表明miR-10b-5p、miR-34b-5p、miR-34b-3p、miR-34c-5p及miR-449a5个miRNAs在精子发生过程中发挥着重要的作用,为课题组后续研究miRNAs在精子发生和男性生育与不育的作用提供研究靶标。NOA相关的差异表达基因的GO和KEGG分析结果提示精子发生过程中的很多环节发生问题都有可能导致非梗阻性无精子症。此外,ASRGL1、SPA4L、LIN7A、RPGRIP1、ZC3H14等基因未见与精子发生的相关研究,可能成为下一步对非梗阻性无精子症相关基因进行功能研究的重点。
