聚天冬氨酸的微波合成及其天然海水阻垢性能*
2022-09-15尹建华王新波
徐 娜,王 玉,徐 旭,尹建华,王新波
(1.自然资源部 天津海水淡化与综合利用研究所,天津 300192;2.山东大学 环境科学与工程学院,山东 青岛 266237)
海水利用是解决我国沿海地区淡水资源短缺的重要途径,受到党和国家的高度重视。根据“全国海水利用报告”统计,截至2012年,我国仅冷却用海水就达到841亿t·a-1。海水为我们提供无尽水源的同时,因其组成复杂性,也带来了严重的结垢和腐蚀问题,造成巨大财产损失甚至安全事故。从安全生产和节约用水等角度考虑,在海水利用过程中,添加水处理药剂进行防垢阻垢处理是必需的。当前,广泛使用的含磷类和聚丙烯酸聚合物类阻垢剂会随排放水进入海洋,造成海水富营养化等环境污染问题[1]。随着人类环保意识的日益增强,对水处理药剂的环保要求也日渐提高,绿色环保型药剂成为21世纪水处理剂发展的方向[2]。聚天冬氨酸(PASP)无毒并具有良好的可生物降解性,对碳酸钙垢有良好阻垢效果,是公认的绿色水处理药剂,已逐渐成为近些年国内外海水阻垢剂研究的热点[3-9]。
目前,制备PASP的工艺路线主要有两条:(1)天冬氨酸直接热缩聚合。以天冬氨酸固体为原料,以酸(主要是H3PO4)为催化剂,以二异丁酮或1,3,5三甲基苯/环丁砜等为溶剂,在180~260℃的高温条件下脱水反应3~5h,PASP的转化率可达到95%左右,该法反应温度高,反应时间长,使用催化剂,会造成能耗大、成本高、污染严重等问题。而且能耗巨大[10-15]。北京化工大学吴一弦等[16]发现采用微波辅助加热,可以在170~200℃的温度条件,20min完成聚合反应,获得高分子量的PASP。但该路线仍然需要有机溶剂和磷酸催化剂,产率也只有40%左右。曹瑞雪等[17]以离子液体为溶剂,微波条件下将收率提高到96.1%,但高成本的离子液体显然会阻碍其大规模生产。(2)氨化聚合法。以马来酸、富马酸、马来酸酐和NH3或铵盐等为原料,共聚后水解生成PASP。该法原料廉价易得,工艺简单,适合大规模工业化生产,但简单加热法制备的产品,相对分子量较低或产率较低[7-10,18]。为提高产率和聚合度,通常需引入催化剂和有机溶剂,则会导致分离过程复杂、增加成本、造成环境污染[10-22]。顾仁杰等[11,23]以马来酸酐和乙酸铵为原料,以NaOH为催化剂,在微波条件下辐射15min,以85%的收率获得PASP,阻垢率达到70%。于跃芹等[12,24]以马来酸酐和NH3·H2O为单体原料,水溶液中微波辐照25min再水解获得PASP,对于自配240mg·L-1的Ca2+溶液,阻垢率达90%。但PASP的产率未被报道,相对分子量只有1100,远低于传统加热法的3000~12000水平。
因此,虽然聚天冬氨酸及其衍生物类阻垢剂的合成已有多条路线被报道,但绿色、高效、高产率和高品质的合成PASP仍然充满挑战。
本文以马来酸酐和NH3·H2O为原料,水为反应介质,在无催化剂,微波辅助加热的条件下,研究了不同操作条件下的聚合反应,以天津汉沽实际海水为测试对象,考察了马来酸酐和NH3·H2O加料比例、微波功率及反应时间等因素对PSI的分子量和产率,以及对PASP海水阻垢性能的影响,确定了最佳的反应条件,构建了反应条件-产物分子量-阻垢性能之间的对应关系。结果表明,无需额外对PASP进行结构衍生,所得产品对实际海水阻垢效率达95%以上,能够满足实际生产需要。
1 实验部分
1.1 试剂与仪器
马来酸酐、NaOH、NaNO3,天津光复精细化工研究所;NH3·H2O、HCl,天津市北方天医化学试剂厂;甲醇、乙醇,天津市津东天正精细化学试剂厂,以上试剂均为分析纯。
EM-310BX型微波炉(三洋SANYO);RO系列5点多点磁力搅拌器、L-MAGHS10型加热磁力搅拌器、EURO STAR型电动搅拌器,广州仪科实验室技术有限公司;TW20型恒温水浴锅(德国Julabo);PHS-3DpH计(上海精密科学仪器有限公司);AL-204型分析天平(梅特勒-托利多仪器有限公司);BL-1200S型电子天平(丹纳赫西特传感工业控制(天津)有限公司);BINDER型真空干燥箱(德国);DHG-9075A型干燥箱(北京雅士林实验设备有限公司)。
1.2 分析方法
凝胶色谱采用美国VISCOTEK公司的Model 302 TDA型凝胶渗透色谱仪,测定温度为30℃,流动相为0.1mol·L-1的NaNO3水溶液,流速1mL·min-1。红外光谱采用美国Nicolet公司生产的Nicolet-6700型傅立叶变换红外光谱仪,仪器分辨率为4cm-1,扫描次数为20,光谱范围为4000~400cm-1。核磁共振色谱仪是Varian公司的400M核磁,型号为UNITYplus,溶剂为D2O。
1.3 中间体聚琥珀酰亚胺(PSI)的合成
马来酸酐和NH3·H2O缩合制备PSI反应式如下:
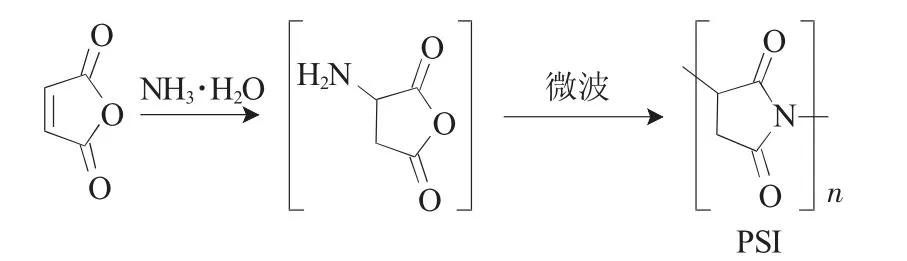
称取一定量的马来酸酐,溶于适量水中,边搅拌边滴加NH3·H2O,加完后继续搅拌5min,然后转移到500mL的烧杯并放入微波炉中;调节微波炉功率和时间,反应得到中间产物聚琥珀酰亚胺粗产品;将粗产物溶于DMF中,抽滤滤去不溶物;取滤液,边搅拌边加适量乙醇,抽滤得到PSI沉淀;将沉淀于80℃下真空干燥12h,得到PSI产物[14,22,23]。产率按式(1)计算:

式中Y:PSI产率,%;m1:PSI的质量,g;m2:投加的马来酸酐的质量,g。
1.4 聚天冬氨酸(PASP)的合成
PSI水解制备PASP的反应式如下:
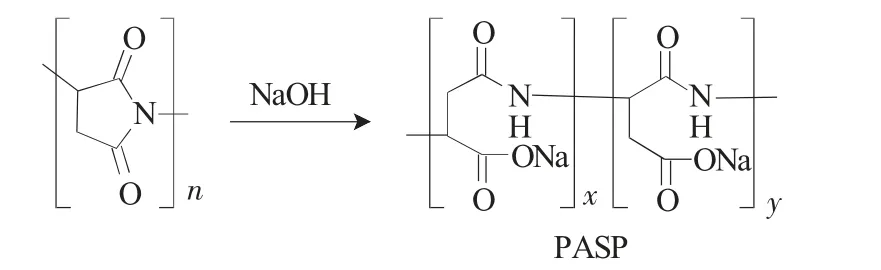
称取一定量的聚琥珀酰亚胺加入2mol·L-1的NaOH溶液于三口瓶中,在50~60℃的水浴条件下搅拌约1h,用2mol·L-1的HCl中和至pH值为6~8。慢慢滴入剧烈搅拌下的甲醇中,抽滤,洗涤,于60℃真空干燥,得PASP[25-28]。
1.5 阻垢率的测定
采用天津汉沽海水,在2倍浓缩条件下进行阻垢率的测定。将制备的1g·L-1PASP阻垢剂,用移液管移取5mL至1000mL烧杯,然后,用容量瓶取1000mL原海水移至烧杯中,将烧杯放入水浴锅,在80℃下蒸发至500mL后盖上盖。恒温24h后,将海水移入500mL容量瓶中,冷却至室温后用蒸馏水定容,用EDTA滴定溶液中的Ca2+,进而算出阻垢率。阻垢率(η)按式(2)计算:

式中η:阻垢率,%;c0:原海水的Ca2+浓度,mg·L-1;c1:未加水处理剂试样实验后的Ca2+浓度,mg·L-1;c2:加水处理剂试样实验后的Ca2+浓度,mg·L-1。
Cl-浓缩倍数与碱度浓缩倍数差值(ΔA)可由式(3)计算:

式中c3:浓缩海水的碱度,mg·L-1;c4:海水的碱度,mg·L-1。
天津汉沽海水的水质参数见表1。
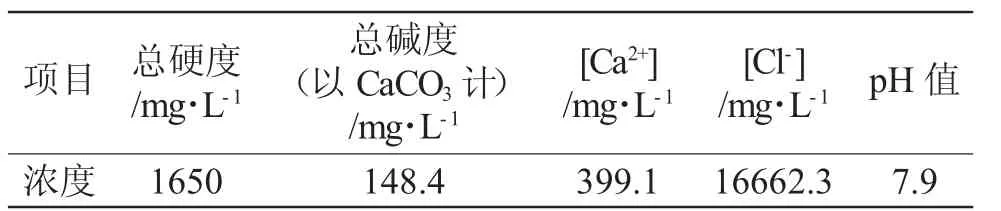
表1 阻垢实验补充水(天津汉沽原海水)水质Tab.1 Scale resistance experiment to replenish the water quality of water(Tianjin Hangu original seawater)
2 结果与讨论
2.1 PSI和PASP的结构表征
微波辅助合成的PSI和PASP纯化后经红外光谱和核磁进行结构表征,结果见图1。
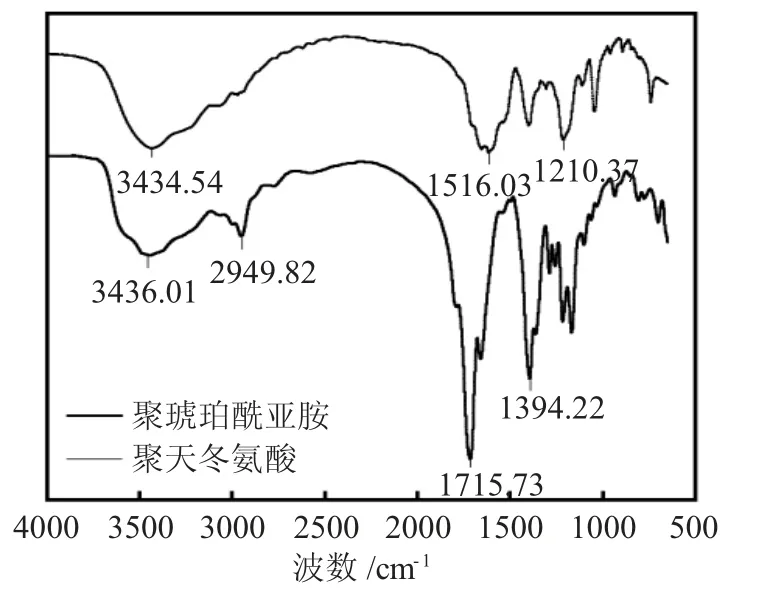
图1 PSI和PASP的红外谱图Fig.1 FTIR spectra of PSI and PASP
由图1可知,PSI的红外光谱与文献一致[9,24],3436.0cm-1处的宽吸收峰表明,结构中存在大量可形成氢键的官能团,可能是酰胺的N-H键,也可能是缩合不完全所残留的羧基等基团;2949.8cm-1处的吸收峰可归属为饱和C-H键的伸缩振动;在1715.7cm-1处出现强吸收峰,说明存在酰胺C=O键的伸缩振动,且由于内酰胺两个羰基的相互耦合作用,在主峰两侧各出现一中等强度吸收肩峰;1394.2cm-1处出现吸收峰,是仲酰胺中-CONH-的N-H键弯曲振动和CN伸缩振动产生的特征吸收,进一步证明了五元环酰亚胺结构被成功合成。经过碱水解后,产物红外光谱在3000~3700cm-1处吸收峰进一步变强变宽,是酰胺键中的-NH-吸收峰和游离羧基的-OH伸缩振动的吸收峰重叠形成;PSI中酰胺C=O键的伸缩振动峰发生明显位移至1615.03cm-1,说明PSI的五元环内酰亚胺发生了水解断裂。
图2为PSI及PASP的核磁谱图。
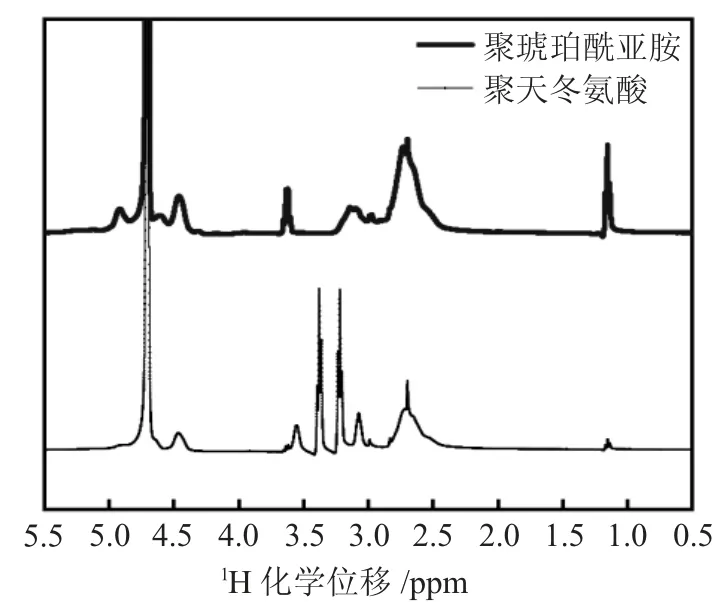
图2 PSI及PASP的核磁谱图Fig.2 1H NMR spectra of PSI and PASP
由图2可知,PSI的1H NMR谱图在δ=2.2~3处出现CH2中的氢核共振峰,在δ=4.3~5处出现羰基邻位CH中的氢核共振峰。相应的,碱水解产物的1H NMR谱图在δ=2.4~3处出现CH2中的氢核共振峰,在δ=3.2~3.4处出现NH中的氢核共振峰,在δ=4.6~5处出现CH中的氢核共振峰。综合红外和核磁测试结果,可以推断PSI和PASP被成功合成。
2.2 合成PSI反应条件的影响
以马来酸酐和NH3·H2O为原料,在无催化剂的情况下,微波法合成了中间产物聚琥珀酰亚胺,详细研究了马来酸酐和NH3·H2O配比、微波功率和微波照射时间对聚琥珀酰亚胺收率的影响,确定了聚琥珀酰亚胺合成的最优反应条件。
2.2.1 正交试验研究 在微波合成聚琥珀酰亚胺的反应中,nNH3·H2O∶n马来酸酐、微波输出功率、反应时间是影响反应的3个主要因素,对聚琥珀酰亚胺的收率有明显的影响。因此,采用三因素三水平正交实验,对聚琥珀酰亚胺的合成反应条件进行了研究。正交实验的因素水平及实验结果分别见表2、3。

表2 正交实验因素水平Tab.2 Orthogonal experimental factor level
由表3可以看出,各因素对聚琥珀酰亚胺收率影响大小的顺序为:A>B>C,即nNH3·H2O∶n马来酸酐>微波输出功率>反应时间。通过正交实验,初步确定的合成条件为:nNH3·H2O∶n马来酸酐=1.4,微波功率700W,反应时间10min。
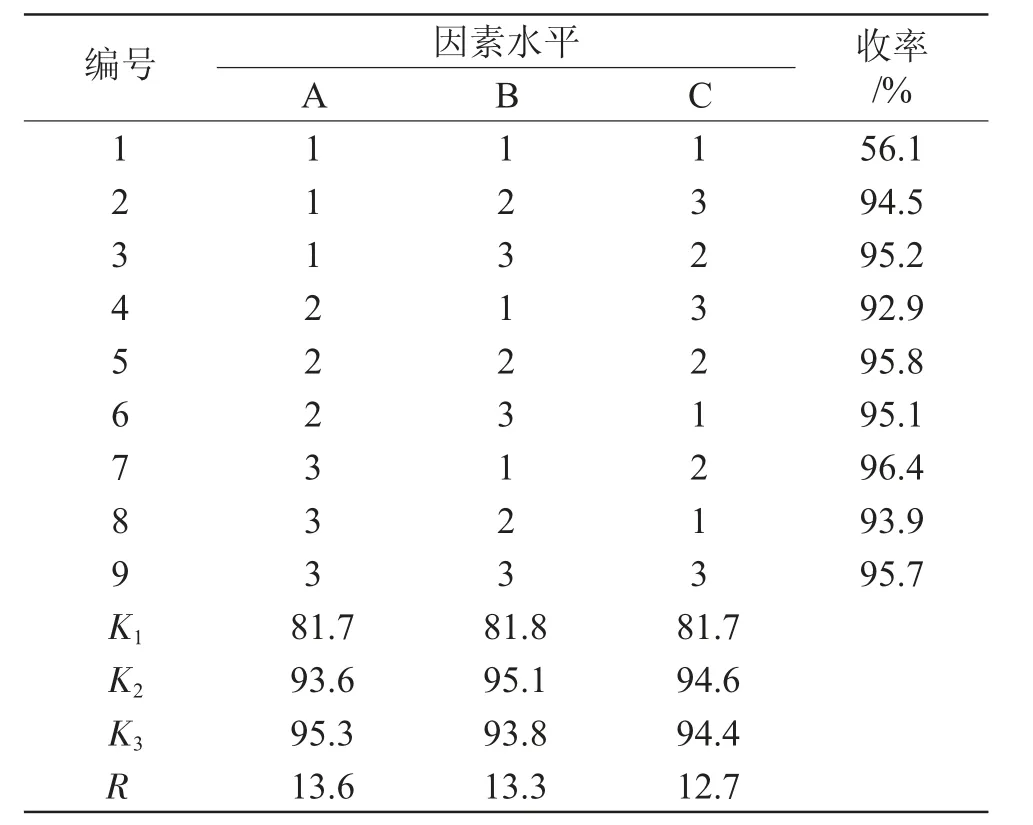
表3 正交实验结果Tab.3 Orthogonal experimental results
2.2.2 NH3·H2O与马来酸酐配比的影响 根据正交试验结果,进一步详细研究了NH3·H2O与马来酸酐配比对PSI合成产率及PASP性能的影响。实验步骤如下:称取马来酸酐9.8g(0.1mol),溶于10g水中;搅拌状态下滴加一定量浓度为26.1%的NH3·H2O,使NH3·H2O与马来酸酐的摩尔比分别为0.6、0.8、1.0、1.2、1.4、1.6、1.8和2.0,反应一段时间后移入500mL烧杯;将烧杯放入微波炉中,调节功率为700W,反应10min。NH3·H2O与马来酸酐配比对聚琥珀酰亚胺收率及聚琥珀酸分子量的影响见图3a。
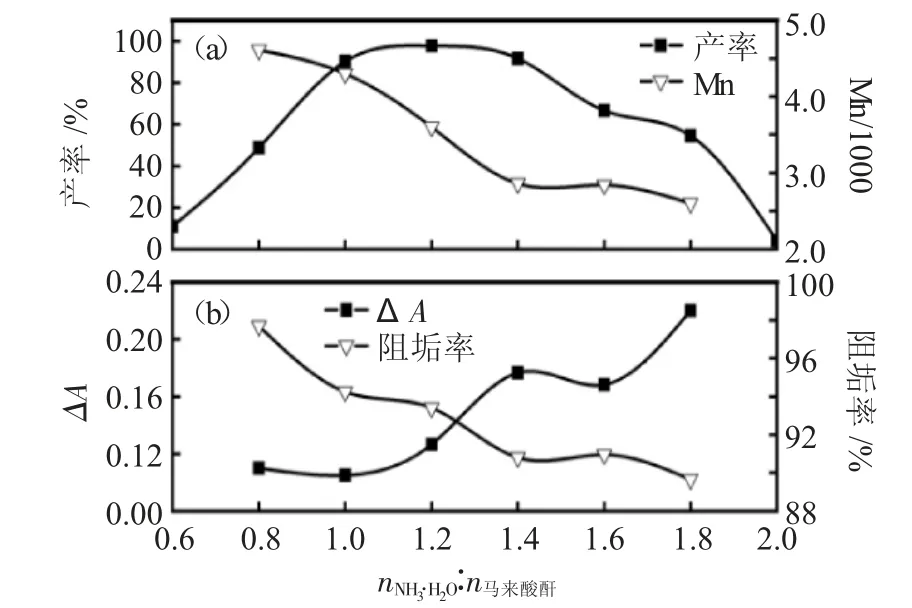
图3 NH3·H2O与马来酸酐配比(a)对PSI收率和PASP分子量的影响;(b)对PASP阻垢性能的影响Fig.3 Effect of ammonia-anhydride ratio(a)on PSI yield and the molecular weight of PASP,(b)on PASP calcium carbonate-inhibiting properties
由图3a中可以看出,随NH3·H2O与马来酸酐配比增大,PSI的收率出现先增大后下降的趋势,当NH3·H2O与马来酸酐配比在1.0~1.4之间时,PSI收率达到90%以上,当配比为1.2时收率达到最高,为95.4%。将合成的PSI水解得到PASP并测定其分子量,结果表明,随nNH3·H2O∶n马来酸酐增大,PASP的分子量呈下降趋势,这可能是由于过多的氨更倾向于形成小分子酰胺化合物,而不是聚合产物。
PASP对汉沽海水的阻垢性能见图3b。对于NH3·H2O与马来酸酐配比在0.8~1.8之间时,PASP阻垢率均可达到90%以上,但整体随比例增大呈下降趋势;ΔA则与之相反,随NH3·H2O添加量的增加呈增大趋势,但在NH3·H2O与马来酸酐配比小于1.8时,ΔA均小于0.2[16],表明微波法合成的聚天冬氨酸具有良好的阻垢性能。阻垢率的变化趋势与PASP分子量变化一致,表明PASP分子量适度增加,有利于提高其阻垢性能。
综合收率、阻垢性能和分子量实验结果,确定NH3·H2O与马来酸酐配比为1.2。这一结果不同于于跃芹等[12,24]的NH3·H2O与马来酸酐最优配比为1.8的结果,说明我们的实验环境下NH3的逸出损失更低,利用率更高。而PASP的分子量从1100提高到了4500,也说明我们改进的反应条件更具优势。2.2.3微波功率的影响 根据正交试验和NH3·H2O/马来酸酐配比实验结果,详细研究了微波功率对PSI合成产率及PASP分子量和阻垢性能的影响。实验步骤如下:称取马来酸酐9.8g(0.1mol),溶于10g水中;搅拌状态下滴加7.8g浓度为26.1%的NH3·H2O,使NH3·H2O与马来酸酐的摩尔比为1.2,反应一段时间后移入500mL烧杯;将烧杯放入微波炉中,分别调功率为1000~150W,反应10min。
随微波功率增大,PSI的收率呈现先增大后下降的趋势,PASP的分子量也呈先增大后减小的趋势,当微波功率为600W时,PSI收率最佳达90%,此时PASP的分子量也恰好是最大的(图4a)。对应PASP的阻碳酸钙垢性能(图4b),除了微波功率为150W的样品,PASP阻垢率均达到90%以上,其中微波功率为600W条件下获得的PASP阻垢率达97%,ΔA为0.08,远小于0.2,表明微波法合成的聚天冬氨酸具有良好的阻垢性能。而高PASP分子量有利于提高其阻垢性能,实验结果与上述原料配比实验中的结论一致。
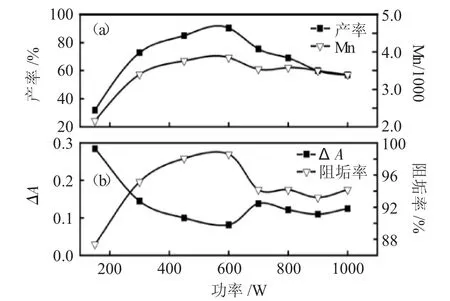
图4 微波功率(a)对PSI收率和PASP分子量的影响;(b)对PASP阻垢性能的影响Fig.4 Effect of microwave power(a)on PSI yield and PASP molecular weight,(b)on PASP calcium carbonateinhibiting properties
综合收率、阻垢性能和分子量实验结果,确定最佳微波功率为600W。
2.2.4 微波反应时间的影响 根据正交实验、NH3·H2O/马来酸酐配比实验和微波功率实验结果,详细研究了反应时间对PSI合成产率及PASP性能的影响。实验步骤同2.2.3,将烧杯放入微波炉中,调功率为600W,控制反应时间分别为4、6、8、10、15、20和25min。结果见图5。
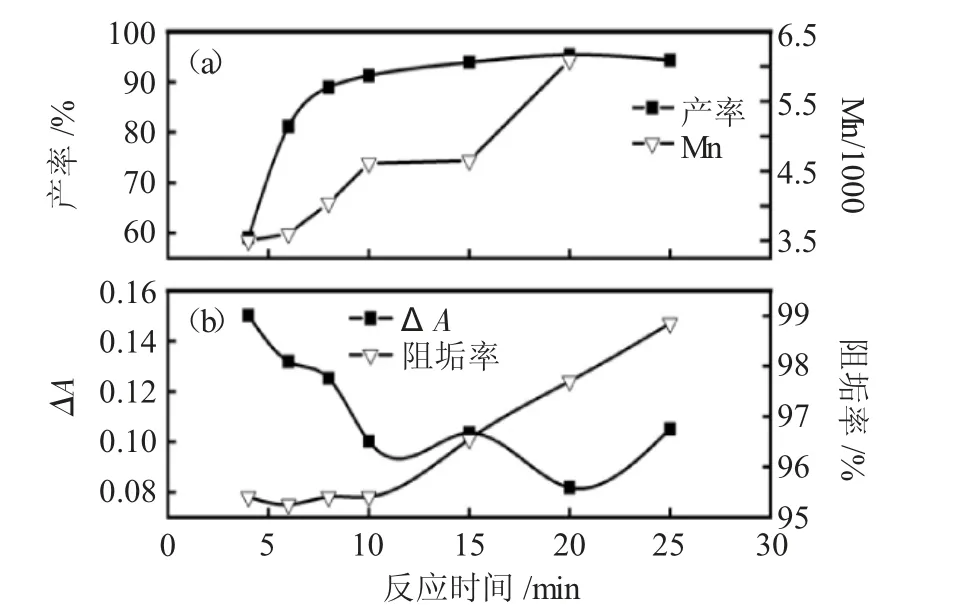
图5 反应时间(a)对PSI收率和PASP分子量的影响;(b)对PASP阻垢性能的影响Fig.5 Effect of reaction time(a)on PSI yield and PASP molecular weight,(b)on PASP calcium carbonateinhibiting properties
由图5a可知,随微波反应时间延长,PSI的收率逐渐增加,在10min后,产率达到91%,继续延长反应时间对收率的增加不明显。将合成的PSI水解得到PASP,测定PASP的阻碳酸钙垢性能(图5b),结果表明,随着初期微波反应时间增长,PASP的阻垢率增大、ΔA减小,但整体均处于比较优异的区间,如PASP阻垢率均达到了94%以上,ΔA均小于0.15。微波反应时间从4min延长至10min,PASP的分子量从3500提高到4600,虽然延长微波反应时间可进一步提高PASP分子量至6300(20min,图5a),但考虑到其阻垢效率已能很好满足需求,延长微波反应时间需要消耗大量能量,并不符合经济性原则,因此,确定反应时间为10min。
3 结论
(1)FT-IR和核磁谱图分析表明,成功合成了聚琥珀酰亚胺及聚天冬氨酸。
(2)通过正交试验和单因素实验相结合的方式,确定了最佳反应条件为:NH3·H2O/马来酸酐配比为1.2、微波功率为600W和反应时间为10min。
(3)在优化的反应条件下,合成PSI的收率可以达到91%,水解后获得的PASP具有高分子量(4610)。以汉沽实际海水为样品的阻垢实验表明,所获得的PASP阻垢剂,能有效阻止海水结垢,阻垢率达到94%,Cl-浓缩倍数与碱度浓缩倍数差值ΔA仅为0.1。
(4)微波辅助合成法相比于传统热缩合方法,在不使用催化剂和有机溶剂的情况下,仍能显著缩短反应时间,提高反应收率和PASP的分子量。
