应用于基因编辑的核糖核蛋白复合体的构建与活性验证
2022-09-14高伟欣黄火清赵晶张鑫杨宁杨浩萌
高伟欣 黄火清 赵晶 张鑫 杨宁 杨浩萌
(中国农业科学院北京畜牧兽医研究所,北京 100193)
近年来,基因编辑技术已经成为生命科学领域最热门的研究工具,其中CRISPR/Cas9基因编辑系统因具有成本低廉、操作简单、编辑效率高等优点,在动物、植物、微生物中都获得了“井喷式”的研究和应用[1-2]。CRISPR/Cas9基因编辑系统可以完成DNA水平上的基因敲除、基因插入、单碱基编辑;并通过与DNA的结合,进行基因的抑制或激活,达到基因表达调控的作用;CRISPR/Cas9系统也可以应用在蛋白质的定向进化、代谢通路的优化、表观遗传学分析、底盘菌株构建等方面,使过去耗时耗力的基因改造过程变得高效,节约了大量的人力、物力和财力[3-7]。
Cas9蛋白的本质是一种DNA内切酶,分子量158 kD左右,目前常用于基因编辑的Cas9蛋白多来源于Streptococcus pyogenes(化脓链球菌)[8]。Cas9蛋白与gRNA(guide RNA)可以在细胞内或体外自发组装成核糖核蛋白复合体(RNP,ribonucleoprotein)。RNP可以“扫描”整个DNA序列,识别出与gRNA上互补的序列,并定位于PAM位点与互补序列上,使DNA双链解开而形成R-loop结构,gRNA与互补链杂交,Cas9蛋白的HNH酶活性位点切割互补的DNA链,而RuvC活性位点将切割非互补链,最终使PAM序列上游3 nt处的DNA双链断裂,完成剪切过程[9-10]。
在对基因组DNA进行基因编辑时,Cas9蛋白和gRNA递送进入细胞的方式一般有3种[11-12]。首先,也是最常用的方式,可以将它们的编码基因克隆在特定质粒上,通过电转、原生质体、农杆菌等转化过程进入细胞,在细胞内完成转录、翻译等过程,最终Cas9蛋白和gRNA结合,发挥基因编辑功能;其次是将编码Cas9蛋白的mRNA和gRNA递送进细胞中,通过翻译合成蛋白并与gRNA结合而发挥功能;也可以将成熟的Cas9蛋白与gRNA一起在体外组装成RNP,然后通过原生质体转化、显微注射等方法导入宿主细胞,同样可以达到切割基因组上特定位点的目的[13]。目前,直接递送RNP的基因编辑已经成功的应用于动物、植物、真菌等的细胞中。例如,在动物基因编辑研究中,对人的T细胞、小鼠的胚胎细胞中与疾病相关基因成功进行了敲除或突变。Wang等[14]研究表明,基于RNP的CRISPR/Cas9基因编辑是研究小鼠免疫系统中基因功能的一种有前景的新策略;Seki等[15]研究表明,采用基于RNP的CRISPR/Cas技术很大程度简化了免疫治疗的基因编辑过程。在植物的基因编辑研究中,如玉米、水稻、烟草等,成功对植物抗病基因进行了改造。Park等[16]应用RNP的基因编辑在较短时间内培育作物的新特性;Khatodia等[17]利用RNP技术使植物具有更持久更广谱的病毒抗性。在里氏木霉、黑曲霉、青霉、稻瘟病菌等真菌的基因编辑研究中,基于RNP的CRISPR/Cas技术促进了酶蛋白、有机酸、抗生素等的生产[18-19]。Kuivanen等[20]研究表明基于体外组装的RNP可以提高靶向效率并且改进了黑曲霉生产半乳酸的能力;Hao等[21]研究证明将Cas9/gRNA复合物转化到里氏木霉中可以实现快速敲除基因的目的,在菌株改良和功能基因组学研究中有广泛的应用。近年来,还发展出基于RNP的DNA内切酶体外活性的作物基因型分析技术、基于RNP的病原体检测技术等,也为RNP的应用提供了新的思路[22-23]。但究竟如何获得有活性的RNP,其详细的体外构建、组装和活性检测的方法及注意事项却很少报导,限制了RNP复合体在基因编辑中的应用。
本研究在大肠杆菌中成功表达了Cas9蛋白,同时,通过体外转录获得敲除目标基因的gRNA,并获得了Cas9蛋白和gRNA自发组装的RNP复合体。经过对RNP复合体的体外活性验证和在黑曲霉中的基因敲除验证,结果表明RNP具有较好的DNA双链剪切活性,在黑曲霉中成功敲除了目标基因,为进一步的菌种改造,提供了良好的材料并大大降低了试验成本。
1 材料与方法
1.1 材料
黑曲霉F223菌株由本实验室保存;表达宿主为大肠杆菌BL21(DE3),pEASY-blunt克隆载体购自北京全式金生物技术有限公司;pET28a(+)由本实验室保存;pREDCas9质粒购自Addgene网站(Addgene #71541;https://www.addgene.org/71541/)。
商品化Cas9蛋白购自GenScript公司;重组酶购自北京全式金生物技术有限公司;硫酸卡那霉素和氨苄青霉素购自索莱宝生物科技有限公司;DNA 胶回收试剂盒购自北京天根生物科技有限公司;DNA高保真聚合酶购自北京全式金生物技术有限公司;限制性核酸内切酶XhoI、SpeI和NcoI购自TaKaRa有限公司;真菌DNA提取试剂盒购自OMEGA公司;UniversAllTMTissue Extraction Kit快速基因组DNA提取试剂盒购自益生生技;T7转录试剂盒MEGAscriptTMT7 High Yield Transcription Kit购自invitrogen公司;RNA纯化试剂盒购自天根生化科技有限公司;蛋白纯化介质为Ni-NTA-agarose resin(Qiagen公司);无填料层析柱购自NEB;蛋白含量测定试剂盒为北京全式金生物技术有限公司产品;蛋白质分子量标准为上海生化研究所产品;其它所用试剂为国产分析纯试剂。
LB液体培养基:0.5%酵母提取物、1%NaCl、1%蛋白胨;固体LB培养基另外加入2%的琼脂粉;YPD液体培养基:2%葡萄糖、2%蛋白胨、0.5%酵母提取物;CD固体培养基(pH 6.0):2%葡萄糖、0.2%氯化钾、0.3%硝酸钠、0.05%七水硫酸镁、0.1%磷酸氢二钾、0.001%七水硫酸亚铁、2%琼脂粉;原生质体转化后生长培养基(pH 7.0):1 mol/L山梨醇、1%葡萄糖、2%琼脂粉;发酵培养基(pH 6.0):5%蔗糖、3%豆粕、2%玉米浆、0.1%硫酸镁。
1.2 方法
1.2.1 载体的构建 以pREDCas9质粒为模板,以Pet-Insert-F和Pet-Insert-R为引物,用高保真聚合酶扩增cas9基因全长序列,并用胶回收试剂盒回收该PCR产物。同时,将pET28a(+)表达载体用XhoI和NcoI做双酶切处理,37℃水浴反应1 h,酶切产物用胶回收试剂盒回收。将以上两个回收片段与重组酶混合,50℃反应15 min,将重组产物转化至大肠杆菌BL21(DE3)感受态细胞,转化方法参考说明书,将感受态细胞在37℃,200 r/min条件下孵育后涂在添加卡那霉素的固体LB平板上,将长出的单克隆进行PCR验证并测序,最终获得包含pET28a-Cas9-NLS表达载体的大肠杆菌BL21菌株。
提取黑曲霉F223的基因组DNA(详见Fungal DNA Mini Kit 说明书),并以此为模板,以glucoamylase-F和glucoamylase-R为引物,高保真聚合酶扩增黑曲霉来源的糖化酶(基因编号:An03g06550)的DNA序列,用胶回收试剂盒回收该PCR产物,克隆到pEASY-blunt载体上,进行DNA测序,获得正确的带有glucoamylase基因的重组质粒,即pEASY-glucoamylase。重组质粒经SpeI酶切线性化,用于Cas9蛋白体外活性验证。
以黑曲霉F223的基因组DNA为模板,以223up-F和223up-R为引物,高保真聚合酶扩增黑曲霉糖化酶上游同源臂;以223down-F和223down-R为引物,同样获得下游同源臂;以hph-F和hph-R为引物,扩增潮霉素抗性基因表达框。用引物223up-F和223down-R将上述3个片段进行融合PCR,胶回收试剂盒回收该PCR产物,获得donor DNA。用引物hph-jc-F和hph-jc-R分别进行测序,验证3个片段是否完成连接。本研究中所使用的引物及序列信息如表1所示。

表1 引物名称及序列信息Table 1 Primer names and sequence information
1.2.2 重组Cas9蛋白的表达 将Cas9蛋白表达菌株活化,按1%接种量转接至200 mL的LB液体培养基中,添加卡那霉素至终浓度为50 μg/mL。将其在37℃条件下,200 r/min摇床培养大约3 h,至OD600=0.6-0.8;加入0.5 mmol/L IPTG诱导蛋白表达,在30℃条件下,200 r/min摇床诱导大约5 h;将菌体在12 000 r/min条件下,离心3 min,用20 mmol/L Tris-HCl(pH 8.0)缓冲液洗一遍菌体,于-20℃冰箱冻存。
1.2.3 重组Cas9蛋白的纯化 冻存的菌体用破碎缓 冲 液(20 mmol/L Tris-HCl,pH 8.0)悬 浮 定 容至20 mL,进行超声破碎,样品参数设置为:功率30%,超声5 s,间隔5 s,总时间为30 min。破碎后在12 000 r/min离心10 min,取上清进行纯化。Cas9蛋白纯化的填料使用Qiagen公司的Ni-NTA-agarose resin,吸取5 mL Ni-NTA-agaroseresin 于空柱子中,注意树脂与垫片间不要留有空隙和气泡。使用含20 mmol/L咪唑的洗涤缓冲液(20 mmol/L Tris-HCl,20 mmol/L imidazole,500 mmol/L NaCl,pH 8.0) 洗5个柱体积,去除杂蛋白,用500 mmol/L咪唑的洗脱缓冲液(20 mmol/L Tris-HCl,500 mmol/L imidazole,500 mmol/L NaCl,pH 8.0)洗脱并收集Cas9蛋白。使用100 000 MW CO蛋白超滤管,将洗脱缓冲液替换为蛋白存储液(20 mmol/L Hepes,pH 7.5,150 mmol/L KCl,1 mmol/L DTT,1 mmol/L EDTA,10%glycerol)。最后使用蛋白含量测定试剂盒对纯化前后的蛋白样品进行定量及SDS-PAGE电泳分析。
1.2.4 gRNA体外转录和纯化 使用E-CRISPR网站(http://www.e-crisp.org/E-CRISP/)选择黑曲霉来源糖化酶基因内部的PAM序列,对gRNA和对照cgRNA进行DNA模板合成(北京博迈德基因技术有限公司)。使用合成的DNA为模板,T7转录试剂盒转录出gRNA和对照cgRNA;具体反应体系如下:DNA模 板200 ng、ATP/UTP/CTP/GTP各4 μL、10× Reaction Buffer 4 μL、Enzyme Mix 4 μL,总 体积40 μL,37℃反应4 h 或过夜,转录后的gRNA和cgRNA稀释100倍,用NanoDrop测定浓度。使用RNA纯化试剂盒对转录获得的gRNA和cgRNA进行纯化,纯化后的产物洗脱体积控制在20 μL,纯化后的gRNA用NanoDrop测定浓度。RNA体外转录模板的序列信息如表2所示。1.2.5 RNP复合体体外活性验证 取纯化后的gRNA(或cgRNA)与Cas9蛋白(或商品化的Cas9蛋白,即cCas9)及线性化的pEASY-glucoamylase质粒DNA混合,反应缓冲液为20 mmol/L Hepes,100 mmol/L NaCl,5 mmol/L MgCl2,0.1 mmol/L EDTA,pH 6.5,反应总体积为20 μL,在37℃条件下反应1 h,通过1%琼脂糖凝胶电泳检测线性化质粒的切割情况。具体试验步骤如下:

表2 RNA体外转录模板的序列信息Table 2 Sequence information of in vitro transcription templates of RNA
首先,设计7个阴性对照试验。分别为线性化质粒DNA;DNA与重组表达的Cas9蛋白;DNA与cCas9蛋 白;DNA与gRNA;DNA与cgRNA;DNA与cgRNA和Cas9蛋白;DNA与cgRNA和cCas9蛋白混合。在37℃条件下反应2 h以上,质粒的添加量都为200 ng,反应总体积为25 μL,通过1%琼脂糖凝胶电泳进行验证。
其次,试验组分为两组,第一组以商品化的Cas9蛋白为试验对象,第二组以纯化的重组Cas9蛋白为试验对象,每组的Cas9蛋白和gRNA的添加量均如表3中所示。反应总体积为25 μL,通过1%琼脂糖凝胶电泳检测线性化质粒的切割情况。

表3 RNP复合体活性验证试验组反应体系Table 3 Reaction system of test group for RNP complex activity verification
1.2.6 RNP复合体体内活性验证 为了验证RNP复合体的体内活性,采用原生质体转化法,将RNP和带潮霉素筛选标记的donor DNA共同转化至黑曲霉糖化酶生产菌株中。黑曲霉原生质体的转化方法参考van等[24]方法。首先将黑曲霉菌丝接种于YPD液体培养基,32℃摇床振荡培养2 d,成为均匀的小球。将其通过目筛过滤取小球,用无菌水冲洗干净后,取适量菌体加入到原生质体酶解液中,30℃,70 r/min荡培养2 h收集原生质体。混合各10 μg的gRNA和纯化得到的Cas9,在37℃条件下孵育30 min,总体积为10 μL,即得到RNP复合体。以PEG介导的原生质体转化法将20 μL回收产物donor DNA和10 μL RNP复合体共同转入黑曲霉中。转化子在潮霉素平板中,32℃培养2-3 d后,挑取转化子至24孔板中,用于阳性转化子的筛选。
首先通过PCR鉴定转化子,用快速提取基因组试剂盒提取基因组DNA,用引物GA223homo-downjc-F和hph-jc-R检测潮霉素基因hph是否被整合到黑曲霉基因组中,筛选阳性转化子。挑取阳性转化子到液体发酵培养基中,在32℃,200 r/min条件下振荡培养5 d后取样。收集上清发酵液,进行SDSPAGE分析。
2 结果
2.1 载体的构建
以pREDCas9质粒为模板,成功扩增了cas9基因全长序列,并在cas9基因3'端通过引物加入了21 bp的核定位信号序列(NLS),该序列编码含7个氨基酸的短肽,能使Cas9蛋白和gRNA复合物(RNP)被识别并通过亲水性的核孔复合物进入细胞核而发挥功能;在核定位信号序列3'端引入了6个His-Tag蛋白纯化标签,便于纯化。将该片段和用XhoI与NcoI进行双酶切处理的pET28a(+)载体进行重组转化,经过测序验证,最终获得了pET28a-Cas9-NLS表达载体。
以黑曲霉F223的基因组DNA为模板,扩增得到了黑曲霉来源糖化酶(基因编号:An03g06550)的DNA序列,获得重组质粒pEASY-glucoamylase,并且经过SpeI酶切成为线性化DNA片段。
以黑曲霉F223的基因组DNA为模板,扩增获得了黑曲霉糖化酶基因上/下游同源臂序列与实验室保存的潮霉素抗性基因表达框进行融合PCR,产物进行测序验证为正确,成功获得了donor DNA。
2.2 重组Cas9蛋白的表达与纯化
经IPTG诱导,细胞破碎和镍柱纯化,对纯化后的Cas9蛋白样品进行SDS-PAGE分析(图1),Cas9蛋白的理论分子量为158 kD,与蛋白电泳中条带位置相符,说明重组Cas9蛋白在大肠杆菌中成功表达,并获得较纯的蛋白样品。Cas9蛋白最终的得率:共8 mL Cas9蛋白洗脱液,测得蛋白浓度约400 μg/mL,原发酵液为200 mL,得率大约为16 mg/L。

图1 重组Cas9蛋白的SDS-PAGE分析Fig.1 SDA-PAGE analysis of recombinant Cas9
2.3 gRNA的转录与纯化
合成的gRNA和对照cgRNA体外转录模板的序列信息见表2。其中特异识别糖化酶基因的20 nt核酸序列用小写字母标注,5'端T7启动子序列为黑体序列,3'端gRNA非识别区序列为斜体序列,将该完整序列进行DNA合成,同时选择另一个在黑曲霉基因组和体外验证用质粒上都不存在的20 nt核酸序列作为对照序列,同样合成对照DNA。
按照T7转录试剂盒操作,获得的gRNA和cgRNA终浓度均为10 μg/μL左右;经RNA纯化试剂盒纯化,gRNA和cgRNA终浓度为5-15 μg/μL左右,该浓度不能太低,否则会导致反应体积过大,剪切效率下降。
2.4 RNP复合体的体外活性验证
如图2-A核酸电泳所示,在7个阴性对照组中,每个反应都在37℃反应2 h以上,都没有发生DNA被剪切的情况,该试验结果表明,Cas9蛋白、gRNA与线性化质粒缺少任何一个,都无法完成剪切反应;另外,因为cgRNA无法识别线性化质粒上的PAM位点,无论其是与重组Cas9蛋白还是商品化的cCas9分别反应时,线性化DNA也都不能被切割。
如图2-B所示,试验组分为两组,第一组以商品化的cCas9蛋白为试验对象,第二组是以纯化的重组Cas9蛋白为试验对象。每个试验组中的Cas9蛋白与gRNA的量按从200 ng到6.4 μg共设了6个反应。当Cas9蛋白与gRNA的量为800 ng时,核酸电泳显示已经有微弱的DNA条带被切割下来,分子量约1.4 kb,与gRNA的识别位点切割DNA后预测大小一致。在随着Cas9蛋白与gRNA的量不断增加时,线性化DNA被切割的更加完全,图中1.4 kb大小的DNA片段的亮度明显增加,未被切割的剩余线性化质粒的分子量也明显降低。由此结果说明,当DNA、Cas9蛋白、gRNA同时存在的情况下,Cas9蛋白与gRNA结合形成RNP,此时Cas9才能发挥剪切作用;另外,随着RNP复合体浓度的提高,剪切效果更加的明显;但是当RNP浓度提高到一定程度时,线性化质粒被完全切割,剪切反应受底物量的限制也会达到平台期,再增加RNP浓度也不会再对质粒的非识别位点进行切割。由此表明,gRNA识别位点的专一性保证了RNP复合体没有发生脱靶切割;在大肠杆菌中表达并且纯化的重组Cas9蛋白与商业化Cas9蛋白具有相似的剪切效果,证明重组Cas9蛋白同样有良好的核酸酶活性。最后,确定了RNP活性检测的最适反应体系为gRNA/Cas9 蛋白的质量各1.6 μg。在25 μL的反应体系中,37℃反应1 h后,可以对200 ng DNA底物充分进行剪切。

图2 RNP复合体体外活性验证Fig.2 In vitro activity verification of RNP complex
2.5 RNP复合体的体内活性验证
对黑曲霉转化子进行PCR验证,检测引物和RNP复合体基因编辑示意见图3。在11个转化子的核酸电泳图4中,与对照原始菌株CK相比,靶向的糖化酶基因内部明显被插入潮霉素抗性标签,获得了特异的1.4 kb左右条带,这些条带经测序验证,潮霉素抗性基因hph成功插入RNP在基因组上的剪切位点内。

图3 RNP复合体体内编辑过程示意图Fig.3 Schematic diagram of RNP complex editing process in vivo

图4 阳性转化子的PCR验证Fig.4 Verification of positive transformants by PCR
对PCR阳性转化子发酵液上清进行SDS-PAGE分析。如图5所示,几乎所有转化子的糖化酶蛋白条带消失,8号转化子与对照菌株相比,糖化酶蛋白条带减弱,说明当将RNP复合物和donor DNA一同转化至黑曲霉中,在RNP复合物的作用下,可以在黑曲霉中实现高效定向整合并敲除目标基因。
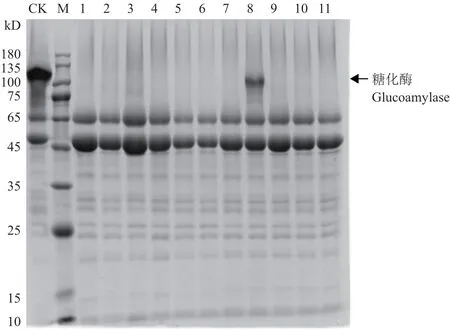
图5 转化子发酵液上清的SDS-PAGE分析Fig.5 SDS-PAGE analysis of supernatant of transformant fermentation broth
3 讨论
RNP复合体介导的基因编辑技术开启了CRISPR/Cas9系统应用的新篇章,成为提高编辑效率、降低脱靶率和免疫反应的有效手段[25]。利用RNP复合体进行基因编辑,不存在外源基因整合进基因组的风险,在细胞内也不会进行RNA的转录和蛋白质的翻译,减少了Cas9蛋白在细胞内停留的时间,减少了Cas9蛋白对宿主细胞的毒性和免疫反应;同时,RNP复合物可以提高gRNA的稳定性,降低了gRNA和Cas9蛋白无法进入同一细胞核的概率,也降低了Cas9蛋白的脱靶率;RNP复合体在完成DNA剪切之后会被细胞内的蛋白酶、核酸酶等降解,在细胞中无残留,可以反复使用RNP转化同一宿主细胞,达到迭代进化的效果;由于gRNA是体外转录的,不受合成量和种类的限制,可以在一次转化过程中使用多个gRNA锚定不同的目标基因,同时进行多个基因的编辑,提高基因编辑效率,加速细胞进化过程[26-27]。
虽然具有以上优势,有剪切活性的RNP复合体的获得却并不简单,在试验设计中需要注意以下几点:(1)Cas9蛋白的基因来源于古细菌,在大肠杆菌中表达需要注意基因中密码子的偏好性。本研究中的cas9基因来源于pREDCas9质粒,该基因已经完成大肠杆菌密码子优化,适于在大肠杆菌中表达,并提高蛋白的表达量;(2)对20 nt的识别位点进行选择时,应该与基因组数据库进行比较,选择在基因组上单一的靶位点,避免基因编辑过程中的脱靶和非靶基因突变,为了提高试验效率,一般选择两个以上的20 nt的识别位点进行试验,从中选择剪切效果好,专一性高的识别位点;(3)gRNA的体外转录使用了T7启动子,保证其转录效率的最短序列为:TAATACGACTCACTATAGGG;(4)本研究用到的gRNA的转录模板DNA为基因合成,成本较高,后期的试验可以利用该模板DNA构建gRNA 转录用“骨架质粒”,不同识别位点的gRNA只需合成一对引物,更换20 nt识别序列即可合成新的gRNA,可以有效节约试验成本。
当然,获得具有活性的RNP复合体后,下一步面临的问题是如何将其递送进宿主细胞。有研究表明,RNP复合体可以通过物理方式,如电穿孔、核转染、显微注射等方法导入细胞,但对细胞有一定的伤害;也可以通过化学方法如聚乙二醇(PEG)介导的原生质体转化、聚乙烯亚胺、树枝状聚合物等介导的转化等方法,但对不同的细胞也会有不同的毒性;另外还有纳米载体介导的RNP复合体递送等其他方法,在动物、植物、微生物、藻类等不同物种中都成功的进行了基因编辑,这些研究成果,为我们的后续试验提供了有效的手段和宝贵的经验[26]。本试验通过采用聚乙二醇(PEG)介导的原生质体的转化方法,将RNP复合体和供体DNA一同转化至黑曲霉中,根据现有实验结果,RNP复合体在体外可以剪切目的基因,在黑曲霉体内也使目的基因表达的相应蛋白条带消失,都说明RNP复合体确实在体外和体内都发挥了基因编辑的作用;通过对转化子的基因组DNA的提取和PCR鉴定,在目的基因上确实整合了潮霉素的筛选标记,与推测的剪切位点和插入位点的PCR条带大小基本一致;我们随机对3个PCR产物进行了测序,确实是在设计的位点上进行了切割和插入,因此体外和体内验证结果一致,都达到了定点剪切的目的,并且基因编辑效率也比较高。
之所以选择黑曲霉作为体内验证实验的受体菌株,是因为黑曲霉作为丝状真菌,是发酵工业的重要生产菌株,已经被广泛应用于生产各种酶制剂和有机酸等[28]。例如,全球市场上60%的柠檬酸是通过黑曲霉发酵生产的;据不完全统计,有30多种工业用酶制剂都可以通过黑曲霉菌株进行工业化生产[29]。利用RNP介导的基因编辑工具对黑曲霉工业菌株进行改造,可以达到提高菌株优化效率的目的,对黑曲霉的工业化应用具有深远的意义。下一步,我们将对RNP在体内基因编辑的条件进行优化,进一步提高编辑效率、降低脱靶率。
4 结论
本研究获得了具有较好剪切活性的RNP复合体,其中纯化的Cas9蛋白具有与商品化Cas9蛋白相似的作用效果,使试验成本大幅度降低;gRNA的转录采用T7转录试剂盒,使gRNA合成的质量得到保证,同时可以针对不同的目标基因合成不同的gRNA,积累成“gRNA库”,为后续试验提供便利。RNP复合体的体外活性检测方法可以在体外直观的检测到核酸剪切的过程,也为RNP复合体在基因编辑之外的基因型分析、病毒检测、活体成像等方面的应用奠定了基础;RNP复合体的体内活性检测,选择工业生产糖化酶的黑曲霉菌株,从SDS-PAGE上直观看到蛋白条带的消失,说明该方法对表达量极高的蛋白也能成功完成基因敲除,为进一步的菌株改造奠定了基础。
