不同健康状态的棘胸蛙肠道菌群结构分析
2022-09-13胡亚洲向建国潘望城
江 昀 胡亚洲 向建国 潘望城
(1. 湖南农业大学动物科学技术学院,长沙 410128;2. 常德大北农饲料有限公司,常德 415400)
棘胸蛙(Paa spinosa,David),又名石蛙、石鸡等,属两栖类无尾目(Anura),主要分布于我国南方山区和越南北部的山林溪涧中,具有较高的营养价值和经济价值[1]。近年来,关于养殖棘胸蛙患歪头病、蓝眼病的报道较多,此类疾病传播速度快、死亡率高和病原菌种类繁多且病因复杂,是蛙类疾病防控的重点[2—7]。
肠道是生物体重要的消化器官,也是重要的黏膜免疫器官,其功能的行使受定植于肠道内的微生物群影响[8]。肠道微生物由常驻菌群和来自食物及外界环境的微生物共同组成,包括古生菌、细菌、真菌和原生动物,其细胞数量和基因数量均远超宿主本身[9]。在哺乳动物中,肠道微生物群落结构与宿主营养、发育、免疫和健康等密切相关[10,11]。水生动物健康与肠道菌群动态平衡高度相关[12,13],病原菌入侵水生动物后,改变其肠道菌群组成并诱导肠道固有免疫的发生[14]。因此了解肠道菌群结构及其与宿主之间的动态相互作用,有助于诊断和治疗与微生物群落失衡相关的疾病[15,16]。
前期研究发现棘胸蛙肠道中定植许多条件致病菌,且发现健康和患病棘胸蛙蝌蚪肠道菌群存在显著差异[1,17],而变态后棘胸蛙的肠道菌群结构与健康状态的关系不得而知。因此本研究基于16S rRNA (16S ribosomal RNA) 高通量测序技术对健康、歪头和蓝眼棘胸蛙的肠道微生物进行调查,分析不同健康状态棘胸蛙肠道菌群结构,旨在为探索棘胸蛙肠道微生物相关疾病的诊断与防治、筛选与肠道健康相关的益生菌等后续研究提供参考。
1 材料与方法
1.1 样本采集
2018年10月8日在湖南省石门县维新镇峡谷河村棘胸蛙养殖场内(110°29′—111°33′E,29°16′—30°08′N,海拔150 m)的5个独立养殖池中采集50个棘胸蛙样本(健康Health 13个,歪头WHD 19个,蓝眼BED 18个)。丁香酚麻醉后,测量样本体长和体重(图1),并在超净台内取出整个肠道于2 mL无菌离心管中,液氮速冻后置于-80℃保存待用。

图1 样本信息图Fig. 1 The information of samples
1.2 基因组DNA的提取和测序
将肠道组织及内容物一起匀浆用于DNA提取。根据DNeasy PowerSoil Kit说明书提取样本的总DNA后,采用Nanodrop 2000分光光度计测定DNA质量。将每个样本总DNA稀释至10 ng/μL后,分为两份对16S rRNA的V4—V5高变区进行PCR扩增。引物使用515F(5′-GTGYCAGCMGCCGCGG TA-3′)和909R(5′-CCCCGYCAATTCMTTTRAGT-3′),其中引物515F的5′端连接12 nt长度的barcod序列用于后续样品分选。
PCR反应体系为: 1×PCR buffer、1.5 mmol/L MgCl2、0.2 mmol/L dNTP、1.0 μmol/L引物、0.25 U ExTaq(TaKaRa,中国)和10 ng DNA模板。PCR反应条件: 94℃预变形3min;94℃变性40s,56℃退火60s,72℃延伸60s,30个循环;最后72℃延伸10min。将同一样本的PCR产物充分混合后,使用1.2%琼脂糖凝胶电泳检测,检测合格后使用San-PrepDNA切胶回收,采用Illumina Miseq进行PE250测序[18]。
1.3 测序处理和质控
将测序所得的下机序列去除低质量、接头污染等数据,用于后续分析。将双端测序的Read1和Read2(分别从5′和3′端测序所得的序列片段)利用FLASH进行拼接,得到 (Merged sequences),用作测序数据量统计。然后用QIIME1.9.0对拼接序列进行筛选,去除与引物不匹配的序列、长度低于300 bp序列、含模糊碱基“N”的序列及平均碱基质量小于30 bp的序列,得到优化序列(Clean sequences)[18—20]。最后去除采用Usearch软件包中的Uchime程序[21]检测到的嵌合体序列,获得有效序列(Effect sequences)[21]。
QIIME1.9.0软件根据序列相似性达到97%进行可操纵分类单元(Operational Taxonomic Units,OTUs)de novo划分,物种注释采用Ribosomal Database Project classifier方法[22],参考数据库为greengenes gg-13-8(http://qiime.org/home_static/dataFiles.html)。
1.4 数据分析
Alpha多样性分析基于本研究获得的总操作分类单元(Operational taxonomic units,OTUs),使用R×64 4.0.2计算各组样品覆盖率(Goods coverage)检验数据的可信度,计算Chao1指数、Shannon指数和Simposn指数检测健康和患病棘胸蛙肠道微生物物种多样性水平。使用GraphPad Prism 8中的单因素ANOVA分析检验健康和患病棘胸蛙肠道微生物多样性指数的差异性。使用R×64 4.0.2绘制Up-Set韦恩图呈现各组样本间OTUs组成的差异性。
Beta多样性分析使用SPSS中的降维分析,将获得的OTUs表进行主成分分析,获取样本对第一、第二主成分的方差贡献率,并利用Adonis计算进行检验。将注释后的OTUs首先进行门分类水平和属分类水平的相对丰度统计。使用SPSS对属水平丰度进行Z标准化,再使用R×64 4.0.2绘制Heatmap图,将数据进行可视化。
2 结果
2.1 16S rRNA测序结果
50个样本共获得1756021条高质量序列,为消除测序深度对后续分析的影响,每个样本随机选取19134条有效序列进行下一步分析。共得到3984个OTUs,35个门,80个纲,155个目,283科,501个属。
2.2 Alpha多样性分析
Alpha多样性指数反映棘胸蛙肠道菌群丰富度和均匀度(图2)。样本覆盖率反映测序深度的合理性,其值越接近1,表明测序深度越合理。本研究中各样本的覆盖率指数除Health组两个样本(>0.978)之外均高于0.98,表明测序深度合理且基本覆盖到样品中的所有微生物群(图2a)。物种丰富度指数在3组之间无显著差异(图2b)。Chao1指数和ACE指数均能反映样本的物种丰富度,本研究中Health组的Chao1指数显著高于WHD组(P<0.01),Health组的ACE指数显著高于BED组和WHD组(P<0.05),而WHD组和BED组之间的Chao1指数和ACE指数均无显著差异(图2e和2f)。Shannon和Simpson多样性指数均能反映物种多样性,Shannon指数更倾向于反映物种丰富度,而Simpson指数更倾向于反映物种均匀度[23],本研究中各组的Shannon指数和Simpson指数之间无显著差异(图2c和2d;P>0.05)。

图2 Alpha多样性组间比较Fig. 2 Alpha diversity of each group
基于OTUs绘制UpSet图(图3),分析3组棘胸蛙肠道菌群多样性关系。结果表明: Health、WHD和BED组总OTUs分别为2453、2413和2361,Health、WHD和BED 3组共享的OTUs数目为1068,占总OTUs的26.8%(图3)。Health组独有24.8%的OTUs,远高于WHD组(8.6%)和BED组(12%;图3)。WHD组和BED组共享OTUs数目达709,在WHD组和BED组中分别占总OTUs的29.4%和30.0%(图3)。WHD组和BED组的OTUs占Health组(OTUs)的百分比分别为12.1%和4.5%(图3)。
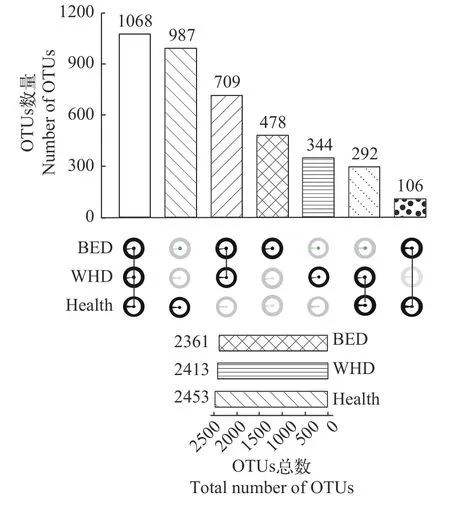
图3 各组的OTUs分析Fig. 3 OTUs analysis of different groups
2.3 肠道菌群Beta多样性分析
为进一步分析各组样本OTUs的差异情况,使用SPSS将获得的OTUs进行主成分分析(Principal Component Analysis,PCA)。根据主成分的特征值(特征值大于1)从样本中共提取到11个主成分,累计贡献率为83.753%,可反应大部分样本信息。提取第一和第二主成分进行后续分析,表明第一主成分主要受到棘胸蛙患病状态的影响,特别是蓝眼症状;第二主成分主要受到棘胸蛙健康状态的影响(图4)。第一主成分的方差贡献率为27.169%,第二主成分的方差贡献率为17.420% (图5)。为进一步分析三组样本的OTUs差异关系,进行Adonis分析,结果表明: 各组间棘胸蛙的OTUs组成差异极显著(P<0.01;表1)。
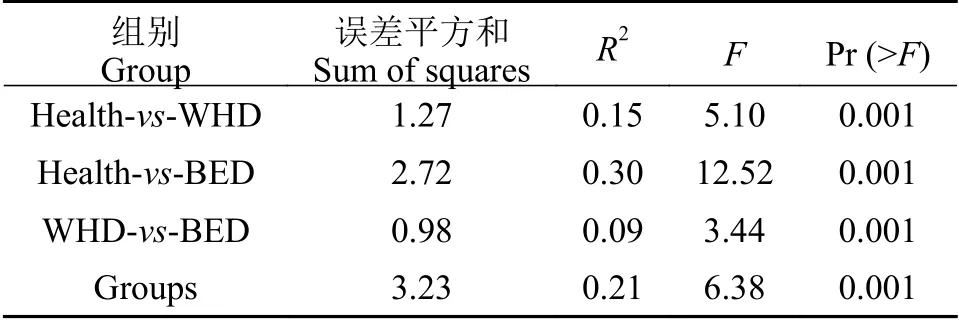
表1 组间Adonis分析Tab. 1 Adonis analysis among groups

图4 各样本在第一和第二主成分的负荷值图Fig. 4 Diagram of load value of samples in PC1 and PC2

图5 PCA分析散点图Fig. 5 PCA Plot
2.4 棘胸蛙肠道菌群结构分析
门分类水平分析基于OTUs组成对Health、WHD和BED组的肠道菌群进行注释,得到相应微生物门类和相对丰度。统计结果显示: Health组的优势菌门是厚壁菌门(Firmicute,52.08% )、拟杆菌门(Bacteroidetes,30.55%)和变形菌门(Proteobacteria,15.55%),其他门类占比不足1%。BED组的优势菌门是拟杆菌门(43.02%)、厚壁菌门(26.48%)、脱铁菌门(Deferribacteres,22.16%)和变形菌门(5.25%),其他门类占比不足1%。WHD组的优势菌门是到厚壁菌门(49.57%)、拟杆菌门(21.93%)、脱铁菌门(14.05%)、变形菌门(10.42%)和柔膜菌门(Tenericutes,1.46%) ,其他门类占比不足1% (图6A)。其中患病组的脱铁菌门大量增加,厚壁菌门、拟杆菌门和变形菌门在患病后丰度均发生明显变化。不同疾病的肠道菌群结构存在显著差异,BED组肠道中拟杆菌门增加,厚壁菌门和变形菌门显著降低(P<0.05);WHD组肠道中厚壁菌门、拟杆菌门和变形菌门丰度均降低(图6B)。

图6 门分类水平上的肠道菌群组成和差异Fig. 6 Intestinal microbiota composition of Paa. spinosa at the phylum level
属分类水平分析筛选3组样本中相对丰度最高的前20种OTUs注释菌属,在属分类水平上进行统计分析。统计结果显示(图7): Health组主要优势菌属为拟杆菌属(Bacteroides,29.70%)、丹毒丝菌科中未定义的一个属(An unidentifiedErysipelotrichaceae,21.38%)、毛螺科中未定义的一个属(An unidentifiedLachnospiraceae,12.23%)、柠檬酸杆菌属(Citrobacter,10.31%)和真杆菌属(Eubacterium,9.09%),其他菌属不足5%;WHD组主要优势菌属为拟杆菌属(16.09%)、毛螺科中未定义的一个属(15.98%)、Mucispirillum(13.89%)、丹毒丝菌科中未定义的一个属(6.92%)和柠檬酸杆菌属(6.19%),其他菌属不足5%;BED组主要优势菌属为Mucispirillum(21.94%)、拟杆菌属(21.76%)和An unidentifiedBacteroidales(7.08%),其他菌属不足5%。其中,拟杆菌属、丹毒丝菌科中未定义的一个属、柠檬酸杆菌属和真杆菌属在患病蛙肠道中丰度呈下降趋势,Mucispirillum在患病组中均大量增加(Health:0.17;WHD: 13.89%;BED21.94%)。

图7 属分类水平上的肠道菌群组成和差异Fig. 7 Intestinal microbiota composition of Paa. spinosa at the genus level
3 讨论
肠道菌群是一个复杂的动态生态系统,参与宿主消化、代谢、调节免疫和抑制致病菌定植等作用[24,25]。影响蛙类肠道菌群结构的因素很多,主要包括宿主因素(如: 物种类型、健康状况和发育阶段)、环境因素(如: 摄食和栖息地类型)、微生物因素(如: 黏附力、酶、生物素和代谢能力)等[17,26,27]。棘胸蛙肠道菌群结构失衡可能诱发疾病,同时不同的健康状态可能也调控棘胸蛙肠道菌群结构[28]。本文基于16S rRNA高通量测序结果分析不同健康状态的棘胸蛙肠道菌群结构,以期探究棘胸蛙健康状态和肠道微生物群落结构之间的相互关系。
3.1 不同健康状态棘胸蛙肠道微生物多样性
Alpha多样性分析表明患病组的ACE指数与健康组之间存在显著性差异(P<0.05),而Shannon指数和Simpson指数无显著性差异(P>0.05)。在棘胸蛙患病后,肠道菌群种类数降低,疾病的发生降低了棘胸蛙肠道菌群的多样性。棘胸蛙肠道是一种特殊的微生态系统,当系统中菌群多样性降低时,系统抵抗外界压力能力降低,其稳定健康的生态环境就容易被破坏[29]。PCA分析和Adonis分析均表明3组样本之间均存在显著性差异,PCA散点图可见健康组和患病组的样本分布在不同区域,但Health组和WHD组样本距离较近,和BED组距离较远,其中WHD组和BED组的部分样本距离相近。另外,疾病组棘胸蛙肠道菌群的多样性发生了明显的变化,健康组和患病组的样本分类聚集,说明棘胸蛙肠道菌群结构与健康状态明显相关。
3.2 不同健康状态棘胸蛙肠道菌门结构与功能分析
厚壁菌门、拟杆菌门和变形菌门是本研究中健康棘胸蛙肠道优势菌群,这与北豹蛙(Lithobates pipiens)[27]、林蛙(Rana dybowskii)[30]、虎纹蛙(Rana rugulosa)[31]和黑斑蛙(Rana nigromaculate)[32]等的肠道菌群相似。我们推测厚壁菌门、拟杆菌门和变形菌门是蛙类肠道中的固有菌群,菌群组成比例受到基因、年龄、性别和养殖环境等因素的影响。物种注释结果显示: 健康组和患病组肠道中的厚壁菌门和拟杆菌门共占肠道菌群近80%,Health组、WHD组和BED组肠道中厚壁菌门和拟杆菌门(F/B)的比值分别是1.71、0.62和2.26。厚壁菌门和拟杆菌门在代谢过程中分别是丁酸盐和丙酸盐的主要生产者,且这两种盐通过调节基因表达,起到抗炎和抗癌的作用[33]。当产丁酸盐的菌群丰度下降后,可能会引起部分条件致病菌的增加[34],这与本研究中BED组厚壁菌门显著降低后,脱铁菌门显著增加的结果一致。棘胸蛙患歪头病后,肠道中F/B比值明显降低,有助于WHD组吸收食物的热量,以补充其在水体中不断旋转游动而消耗的大量能量[25,35]。我们推测棘胸蛙肠道中F/B比值越小,吸收和储存能量的潜力可能越强[34,36]。棘胸蛙肠道的F/B比值明显增大,动物性营养的消化代谢水平降低,丙酸盐代谢增强,在一定水平上增强宿主的饱腹感,以应对视力降低导致摄食降低的情况[35]。变形菌门广泛参与宿主的营养代谢,其丰度受拟杆菌门和厚壁菌门的总丰度影响[37]。门分类水平分析发现,WHD组和BED组肠道中的脱铁菌门丰度显著上升,分别约是Health组的123倍和78倍。脱铁菌是一类专性或兼性厌氧菌,参与硫化合物和铁循环,可还原硫、铁(iii)、硝酸盐和砷酸盐[38,39]。肠道菌群中脱铁菌门的铁代谢与肠道铁平衡密切相关,铁代谢异常不仅增加患病风险,还会促进肿瘤生长[40]。此外LeÓn等[41]。研究发现,小鼠结肠黏膜中的促炎细胞因子Ⅱ-1β的基因表达与脱铁菌的上调成正相关因此我们推测WHD组和BED组肠道中脱铁菌门丰度的显著上升(P<0.05)可能与患病棘胸蛙的营养代谢、解毒及免疫应答过程有关。
3.3 不同健康状态棘胸蛙肠道菌属结构与功能分析
拟杆菌门中的拟杆菌属和柠檬酸杆菌属共占健康蛙肠道菌群的40.01%,厚壁菌门中的丹毒丝菌科中未定义的一个属、毛螺科中未定义的一个属和真杆菌属占健康蛙肠道菌群的42.70%,拟杆菌门中主要菌属与厚壁菌门中的主要菌属的比值为0.94。患病后,WHD组和BED组的比值分别为0.83和2.31,该结果和门分类水平中厚壁菌门和拟杆菌门的比值正相关。我们推测棘胸蛙患病后,这几类优势菌属对门水平丰度的变化起着重要作用。拟杆菌属和柠檬酸杆菌属与宿主营养、免疫、生长发育、代谢和肿瘤的发生等相关[42—44]。丹毒丝菌和毛螺科在人和小鼠的肠道炎症中起着相反的作用。丹毒丝菌科与TNF因子的升高相关,能够促进炎症的发生;而毛螺菌科是短链脂肪酸的主要生产者,可防止过度炎症反应[45,46]。毛螺科、真杆菌属和梭菌属是丁酸盐的重要生产者,可增强宿主的抗炎水平[25,47]。我们推测棘胸蛙患病后,肠道中产短链脂肪酸(如丁酸盐、丙酸盐等)的菌群大量增殖后,抑制了丹毒丝菌科的增殖。
Mucispirillum属脱铁菌门,是一种定植于宿主肠道中的革兰氏阴性菌属,在肠炎时大量增殖,可穿过肠黏膜屏障引起宿主免疫反应[48]。以往对肠炎小鼠研究发现,Mucispirillum与肠炎及炎症标志物相关,其中Mucispirillum schaedleri定植小鼠肠道后参与宿主炎症过程中的氧化应激反应。且该菌属含有的Ⅵ型分泌系统和效应蛋白,能够通过改变宿主的黏膜基因表达来增强宿主的炎症反应[49]。基于属水平分析发现WHD组和BED组肠道中的Mucispirillum分别是Health组的81.7和129.1倍,而引起棘胸蛙歪头和蓝眼病的病原为伊丽莎白菌属,因此我们推测棘胸蛙患歪头和蓝眼病后引起肠道炎症的发生,Mucispirillum大量增殖[5]。
4 结论
动物肠道是重要的免疫屏障,其免疫功能与肠道菌群密切相关,因此构建良好的宿主肠道-微生物生态系统有助于宿主健康。我们研究发现,厚壁菌门、拟杆菌门和变形菌门是健康棘胸蛙肠道微生物的优势菌门,脱铁菌门中的Mucispirillum在患病棘胸蛙肠道中高度富集,这些结果表明健康状态与棘胸蛙肠道菌群结构与组成密切关联。然而是否因健康状态改变引起肠道菌群失调,还是肠道菌群失调引起健康状态改变,及本研究中检测到的健康和患病棘胸蛙肠道中差异显著的菌属是如何参与棘胸蛙肠道免疫功能,这些疑问均有待进一步研究。
