先天性肝纤维化合并常染色体显性遗传性多囊肾病家系分析
2022-09-13郑玲玲刘南南马珺珺孟银梅叶小欣张杰灵李卫
郑玲玲 刘南南 马珺珺 孟银梅 叶小欣 张杰灵 李卫
患者,女性,40岁,因“反复乏力、纳差3年余 ”于2019年入院,既往有多囊肾病史7年,患者3年前无明显诱因下反复出现乏力、纳差,在当地医院发现血小板降低并诊断为原因不明肝硬化。配偶健康,否认家族近亲婚育史。患者父亲36岁时因肝癌去世;哥哥17岁时因肝硬化上消化道出血去世,有多囊肾病史;双胞胎妹妹36岁时发现不明原因肝硬化失代偿期且有多囊肾、多囊肝病史,患者儿子有多囊肾病史。体格检查:神志清楚,全身皮肤黏膜正常,心肺无明显异常。腹平坦,腹部未见静脉曲张,腹软,全腹无压痛,无反跳痛,肝肋缘下不能触及,脾肋下3 cm可触及,胆囊不能触及,墨菲征阴性,移动性浊音(-),双下肢无水肿。辅助检查:血常规白细胞2.24×109/L,红细胞4.1×1012/L,血红蛋白107 g/L,血小板63×109/L;甲状腺功能:FT3 2.66 pg/mL,FT4 1.25 ng/dL,TSH 9.47 IU/mL,肝、肾功能、心肌酶谱、电解质、凝血功能五项、尿粪常规、甲胎蛋白均未见异常;常见肝炎病毒检测、EB病毒、巨细胞病毒等均为阴性,免疫球蛋白、转铁蛋白、铜蓝蛋白、抗核抗体、抗线粒体抗体、抗平滑肌抗体等均未见异常;12导联心电图:室早二联律;胸部X片正常;24 h动态心电图:频发室早二联律,短阵室性二联律。心脏彩超:左房增大,二尖瓣少量返流。上消化道造影(钡餐)示胃炎。头颅磁共振血管成像未见明显异常。FibroScan:CAP 226 dB/m,E 27.4 kPa。上腹部MRI平扫+增强(图1):肝内脉管系统紊乱,肝内外胆管未见明显扩张,脾脏体积增大,肝脾周围见少量液性信号,双肾见多发类圆形长T1T2囊性信号影。肝硬化、脾大,少量腹水及多囊肾。肝组织病理报告(图2):光镜可见汇管区扩大,宽大致密纤维瘢痕形成,胆管畸形,分支增多,可见胆栓形成,肝细胞区域性水样变性,少量点灶状坏死。病理诊断:先天性肝纤维化;免疫组化:HBsAg(-),HBcAg(-),mum-1(个别+),CK7/CK9示胆管阳性。特殊染色:铜(-),铁(-)。家族史:患者有肝硬化和多囊肾病家族史,遂对患者及亲属进行了详细的家系调查及常规检测,发现患者即此家系的先证者,其儿子、哥哥、姐姐、双胞胎妹妹及妹妹的1子1女均发现有多囊肾病史,符合ADPKD遗传规律(图3)。随后对先证者的母亲、姐妹及子女分别采集外周血标本提取DNA进行全外显子组测序分析(广州欧蒙未一医学检验实验室),结果显示多囊肾病1型致病基因(polycystic kidney disease 1 gene,PKD1)出现NM 001009944.3:c.5086C>T位点杂合性突变。后续经Sanger测序法确认先证者、先证者儿子、先证者姐姐、双胞胎妹妹及妹妹的子女体细胞中均存在同一位点杂合性突变。但先证者母亲、先证者姐姐的子女相同位点均为野生型(图4)。将本病例检测到的突变位点与ADPKD突变数据库(Autosomal Dominant Polycystic Kidney Disease Mutation Database,PKDB;http://pkdb.mayo.edu/)进行比对,确认为PKD1的已知致病突变位点。诊断:CHF合并ADPKD;患者的姐姐、患者的儿子、姐妹的子女均暂未发现肝功能异常。予以患者对症支持治疗后好转出院,随访至2021年6月,仅患者双胞胎妹妹因肝硬化腹水低蛋白血症曾在当地医院住院治疗,患者及其他家人病情均稳定。
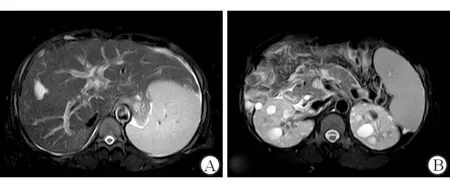
A:肝内脉管系统紊乱;B:脾大,双肾多发囊肿
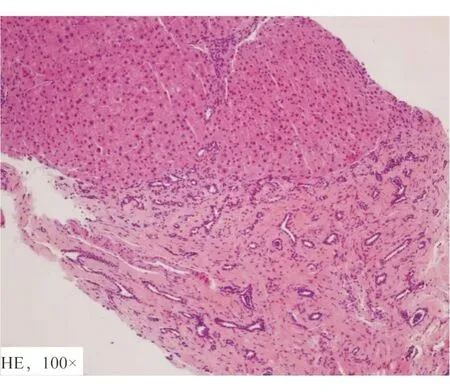
图2 肝脏组织病理检查结果
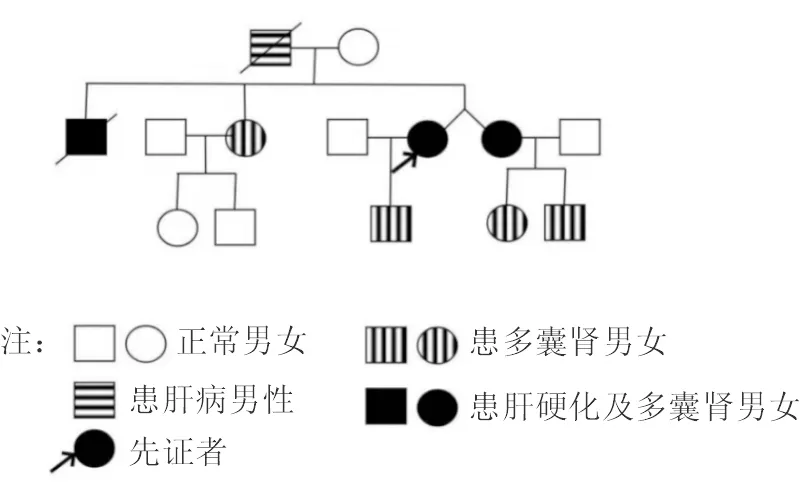
图3 患者家系图谱
讨论先天性肝纤维化(congenital hepatic fibrosis,CHF)是一种少见的与胆管板畸形有关的常染色体隐性的遗传病,可单独发病,也可合并多囊肾(polycystic kidney disease,PKD)、先天性肝内胆管扩张症(Caroli病)。国内文献报道的CHF多合并常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)和 Caroli病[1],而合并常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)则罕见有报道。主要表现为门静脉高压及相关并发症和复发性的胆管炎,而肝细胞功能正常或轻度异常但没有特异性,影像学、病理检查和遗传学评价对其诊断很重要,病理活检是诊断金标准[2]。本例患者有肝硬化及门脉高压的表现,肝穿刺活检证实先天性肝纤维化,诊断明确,但患者的妹妹在当地因肝硬化失代偿期,血小板极低未能行肝穿刺活检。
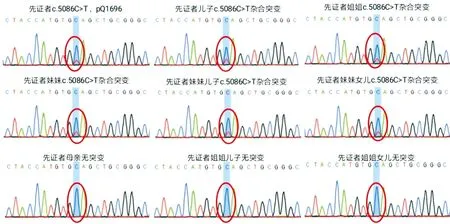
图4 PKD1基因测序结果
多囊肾病是临床常见的遗传病之一,常见ARPKD和ADPKD两种类型。CHF与ARPKD和ADPKD都属于遗传性纤维囊性疾病,但CHF多发生在ARPKD中,目前研究认为,CHF、ARPKD和Caroli 病均与多囊肾/多囊肝病变1基因(polycystic kidney and hepatic disease 1 gene,PKHD1)基因突变有关[3],因此CHF合并ARPKD临床多有报道,但CHF发生在ADPKD罕见。临床上ADPKD要比ARPKD多见,其主要致病基因为PKD1和多囊肾病 2 型致病基因(polycystic kidney disease 2 gene,PKD2)[4],可累及全身器官,有肾脏表现和肾外表现,多为成年后发现双侧肾囊肿并随年龄增长出现肾脏结构及功能异常,还可出现肝脏、胰腺、脾脏、精囊囊肿、心脏瓣膜病和颅内动脉瘤等,当同时满足主要诊断标准和任何一个次要诊断标准即可临床诊断为ADPKD,主要诊断标准包括明确家族史且符合常染色体显性遗传和双肾多发液性囊肿,次要标准包括多囊肝、腹部疝、肾功能不全、精囊囊肿、心脏瓣膜异常和颅内动脉瘤。基因检测也可辅助诊断,尤其适用于无明确家族史者,或是影像学不典型等特殊ADPKD病情[4]。
本病主要与ARPKD进行鉴别诊断,ARPKD是多发于儿童的常染色体隐性遗传病,以两肾多发进行性增大的囊肿及肝门脉系统发育不全为特征,致病基因为PKHD1,与ADPKD最重要的鉴别是没有父母肾囊肿的家族史[5]。有研究提示PKD1基因突变型预后较PKD2差,病情也更重,另外早期出现高血压、血尿、蛋白尿、肾体积增大均会使ADPKD预后变差[4]。本例患者及患者儿子均有致病基因,患者母亲没有致病基因,结合患者父亲肝癌去世已久及ADPKD的显性遗传规律,考虑突变位点来自患者父亲,患者有多囊肾家族病史,且符合常染色体显性遗传规律,有肾脏影像学表现,基因检测也证实有已知致病突变基因,诊断明确,结合家系PKD1基因突变预后更差,密切随访,积极对症治疗,延缓进入终末期肾病。
目前尚无有效治疗可以阻止或逆转CHF进程,主要早期干预门静脉高压和感染,控制远期并发症,延缓病变的进展,肝移植为目前CHF有效的根治方法[6];对于PKD目前也无明显特效治疗,主要积极控制并发症和对症治疗,血管加压素2受体拮抗剂(托伐普坦)可用于减少控制囊肿的生长[3];明显肝肾受累的患者可进行肝肾联合移植手术[7]。对于下一代,建议ADPKD患者进行遗传咨询,可考虑利用基因测序及胚胎植入前遗传学技术从根本上阻断遗传,降低患儿出生率[4]。本例患者随访中病情稳定,但患者妹妹处于肝硬化失代偿期,建议肝移植治疗。
在临床工作中不明原因肝硬化并不少见,而多囊肾病也是常见肾病之一,有些家族史并不明确,旨在通过本例患者报道能提高医务人员对CHF伴PKD疾病的认识,早期快速识别患者,由于本病无有效治疗方法,远期预后差,且是遗传病,对整个家族健康造成极大损害及负担,所以提高早期检出率对患者预后及整个家族健康至关重要。
